 图式1
单体与聚合物的合成路线.
图式1.
Synthetic route of monomers and polymers
图式1
单体与聚合物的合成路线.
图式1.
Synthetic route of monomers and polymers
引用本文:
贾涛, 郑楠楠, 蔡万清, 应磊, 黄飞. 基于萘并二酰亚胺的胺基功能化聚合物的三组分一锅法合成及其在聚合物太阳电池中的应用[J]. 化学学报,
2017, 75(8): 808-818.
doi:
10.6023/A17030114

Citation: Jia Tao, Zheng Nannan, Cai Wanqing, Ying Lei, Huang Fei. Naphthalene Diimide-Based Polymers Consisting of Amino Alkyl Side Groups:Three-Component One-Pot Polymerization and Their Application in Polymer Solar Cells[J]. Acta Chimica Sinica, 2017, 75(8): 808-818. doi: 10.6023/A17030114
Citation: Jia Tao, Zheng Nannan, Cai Wanqing, Ying Lei, Huang Fei. Naphthalene Diimide-Based Polymers Consisting of Amino Alkyl Side Groups:Three-Component One-Pot Polymerization and Their Application in Polymer Solar Cells[J]. Acta Chimica Sinica, 2017, 75(8): 808-818. doi: 10.6023/A17030114
基于萘并二酰亚胺的胺基功能化聚合物的三组分一锅法合成及其在聚合物太阳电池中的应用
摘要:
通过微波辅助炔、醛、胺三组分一锅法聚合反应合成了一系列基于萘并二酰亚胺的聚合物P1~P4,并通过核磁表征确认其结构.目标聚合物的光物理与电化学性能研究表明,由于聚合物主链共轭被打断,聚合物的吸收和能级主要由其重复单元决定.通过开尔文探针与电子顺磁共振谱研究了聚合物侧链与主链胺基不同的化学环境对其电极功函调节以及自掺杂行为的影响.发现由于主链胺基与苄基、炔丙基相连,胺基氮的电子云密度显著降低,导致聚合物形成界面偶极能力减弱,从而使其电极功函调节能力与自掺杂强度都大大降低.所有聚合物中P1具有良好的醇溶性,可被用于聚合物太阳电池的阴极界面层.以PTB7-Th:PC71BM为活性层,P1为阴极界面层的聚合物太阳电池器件的光电转换效率达到9.34%.
English
Naphthalene Diimide-Based Polymers Consisting of Amino Alkyl Side Groups:Three-Component One-Pot Polymerization and Their Application in Polymer Solar Cells
Abstract:
In this work, we demonstrate the microwave-assisted synthesis of naphthalene diimide-based polymers via three-component polymerization (TCP) of diynes, dialdehydes and dibenzylamine, and the applications of such polymers as cathode interfacial layers for polymer solar cells. The TCP of diynes (1a~1c), dialdehydes (2a~2b) and dibenzylamine catalyzed by InCl3 could be performed smoothly under microwave irradiation in very short reaction time, yielding soluble polymers P1~P4 with high molecular weights. The chemical structures of these resulting polymers were confirmed by nuclear magnetic resonance spectroscopy. The thermal stability, photophysical and electrochemical properties of the resulting polymers were also investigated. Besides, the effects of chemical environment of amine groups on the resulting polymers' electrode modification capability and self-doping behavior were explored by conducting scanning Kelvin probe microscopy and electron paramagnetic resonance (EPR) spectroscopy studies, respectively. It was found that the chemical environment variation of amine groups, including the decreasing electron density of the nitrogen atoms in alkylamine and the enhancing steric hindrance around the nitrogen atoms from substituent groups, can substantially influence the electrode modification capability and self-doping behavior of the resulting polymers. Moreover, quantum chemistry calculation was also conducted to qualitatively illuminate the essential distinction in chemical environment of different amine groups. It was found that the negative atomic dipole moment corrected Hirshfeld (ADCH) charge of nitrogen atoms in side chains was significantly larger than the ADCH charges of nitrogen atoms in main chains. Among all the resulting polymers, P1 can be easily dissolved in alcohol due to its amino functionalized side chain groups and thus was utilized as the cathode interlayer for polymer solar cells. The device with P1 as the cathode interlayer and PTB7-Th:PC71BM as the photoactive layer exhibits a high power conversion efficiency of 9.34%, which is much better than that of the control device without such cathode interlayer. All these results provide a guideline for the material design of amino-functionalized polymers for the optoelectronic devices. And it was also shown that the multicomponent polymerization (MCP) is an effective strategy for the synthesis of functional polymers, and may trigger broad research interests in developing effective polymerization approaches toward multi-functional polymer materials.
-
Key words:
- three-component polymerization
- / polymer solar cells
- / cathode interlayer
-
1 引言
多组分反应是将三种或三种以上反应物混合到一个反应体系, 无需对中间产物进行分离提纯, 直接得到含所有底物结构片段终产物的化学反应[1, 2].相比传统的多步合成方法, 多组分反应操作简单、反应高效、选择性好、且具有原子经济性, 因而广受有机合成化学家的青睐[3~10].多组分反应与组合化学相结合, 被广泛用于天然产物的合成及新药开发等领域, 成为合成多样性复杂化合物的有效策略[11~15].同时, 功能聚合物的广泛研究与应用也激励化学家们将种类繁多的多组分反应扩展到聚合反应领域, 制备结构确定, 单元序列可控的多功能化聚合物.近年来, 困扰这类反应的底物适用性、产物分子量、溶解性及副反应等问题不断得到解决[16~18].例如, Meier及其课题组[19]通过Passerini多组分反应制备了单元序列确定的聚酰胺; Choi等[20]报道了Cu催化多组分聚合制备高分子量、无结构缺陷的聚磺酰亚胺; 唐课题组[21~26]则报道了一系列基于三键的多组分聚合反应.尽管多组分聚合承继多组分反应的诸多优点, 但多数聚合反应还停留在合成探索阶段, 功能化应用实例较少.因此, 通过多组分聚合反应制备功能化聚合物, 并扩展挖掘其应用价值至关重要.
近年来, 聚合物太阳电池(PSCs)因其质轻、柔性、低成本等优点受到广泛关注[27~31].高效聚合物太阳电池的关键除了高性能的活性层材料外[32~34], 优异的界面材料也不可或缺[35, 36].胺基功能化聚合物如聚[(9, 9-双(3'-(N, N-二甲胺基)丙基)-2, 7-芴)-交替-2, 7-(9, 9-二辛基芴)] (PFN)等作为阴极界面层, 已被成功应用于聚合物太阳电池[37~39].其中, 极性胺基侧链不仅确保聚合物具有醇溶性, 解决了正交溶剂加工制备多层器件的问题, 同时还使聚合物具有一系列新功能:可调节电极的功函数, 提高电子抽提效率; 可在界面处掺杂富勒烯, 提升器件短路电流; 胺基可充当空穴陷阱, 减少界面处双分子复合; 诱导活性层形成垂直相分离结构, 改善载流子传输等[40~42].已有文献报道胺基功能化聚合物侧链富电性的氮原子对偶极形成与电极功函调节至关重要[43, 44], 然而之前对胺基功能化聚合物的研究多数集中在对主链[45, 46]或侧链[47, 48]功能单元的调节.为进一步深入理解胺基功能化聚合物的界面作用机理以及结构-性能的关系, 阐明胺基所处的化学环境对材料性能的影响具有重要意义.
在之前的工作中, 我们成功地通过InCl3催化二炔、二醛、二苄胺三组分聚合反应制备了胺基功能化的聚合物, 并将其用于聚合物太阳电池的阴极界面层, 展现了三组分聚合反应在制备多功能聚合物方面的潜在应用[49].萘并二酰亚胺(NDI)单元具有优异的电子传输性能, 因而被广泛应用于有机场效应晶体管[50, 51]与全聚合物太阳电池[52~54]材料体系.因此, 本文将NDI单元引入到多组分单体中, 合成一系列的多组分单体二炔(1a~1c), 二醛(2a~2b) (Scheme 1), 并通过微波辅助三组分一锅法聚合反应制备了多组分聚合物P1~P4.此外, 聚合反应中原位生成的与侧链胺基化学环境不同的叔胺, 为我们研究胺基化学环境不同对材料界面修饰功能以及自掺杂现象的影响提供了可能.研究结果表明三组分聚合获得的聚合物具有较好的热稳定性与溶解性, 由于主链共轭被打断, 聚合物的吸收和分子轨道能级主要取决于重复单元的吸收及能级.进一步通过开尔文探针与电子顺磁共振谱研究了聚合物侧链与主链胺基化学环境不同对材料功函调节、自掺杂行为的影响.结果表明, 一方面聚合物主链胺基与苄基、炔丙基相连, 使其胺基氮的电子云密度显著降低, 另一方面取代基的位阻效应还可能阻碍氮上孤对电子与金属电极的作用.这两方面的协同作用使相关聚合物的电极功函调节以及自掺杂能力都大大减弱.在所有聚合物中, 侧链含有胺基的聚合物P1具有良好的醇溶性, 可以作为阴极界面层应用于聚合物太阳电池中.以P1为阴极界面层, PTB7-Th:PC71BM为活性层制备的聚合物太阳电池光电转换效率达到9.34%.
图式1
单体与聚合物的合成路线.
图式1.
Synthetic route of monomers and polymers
2 结果与讨论
2.1 聚合物的合成与表征
微波辅助三组分聚合反应条件参考之前文献报道[49], 通过优化反应时间与温度, 聚合反应得到了相对分子量高、溶解性良好的聚合物.如图 1所示, 二苄胺、单体1b、单体2b以及目标聚合物P2的1H NMR对照图.化学位移δ 3.81, 3.12与10.14分别为二苄胺的亚甲基氢、单体1b炔基氢、单体2b醛基氢, 这些特征化学位移峰在聚合物P2中消失.与此同时, 化学位移δ 5.12, 3.93与3.68出现了新的核磁峰.化学位移δ 5.12为新生成的炔丙基上的氢, 化学位移δ 3.93与3.68为二苄胺的亚甲基氢.此外, 聚合物P2碳谱中没有出现单体2b羰基碳δ 191.7的化学位移峰, 单体1b炔基碳化学位移则从δ 84.5与74.4分别移向δ 91.4与84.9, 二苄胺亚甲基碳的化学位移则出现在δ 56.2与54.9.这些核磁表征的结果表明我们得到了目标聚合物.此外, 所得聚合物在常见溶剂四氢呋喃、甲苯、氯仿、氯苯中具有较好的溶解性. P1~P4中, P1因为侧链胺基的存在而能溶于甲醇, 所以可通过正交溶剂加工制备多层电子器件.通过热重分析仪对聚合物的热稳定性进行了表征(图 2).结果表明, 目标聚合物都有较好的热稳定性, 热分解温度在270 ℃左右.
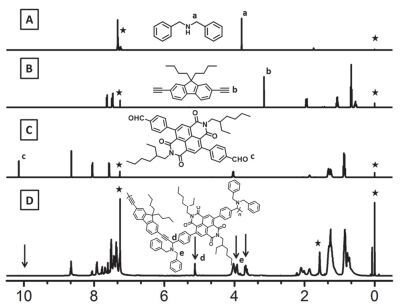 图 1
(A)二苄胺, (B) 1b, (C) 2b, 聚合物P2在氘代氯仿的核磁图.溶剂峰以五角星标出
Figure 1.
1H NMR spectra of (A) dibenzylamine, (B) 1b, (C) 2b and (D) polymer P2 in deuterated chloroform (the solvent peaks are marked with asterisks)
图 1
(A)二苄胺, (B) 1b, (C) 2b, 聚合物P2在氘代氯仿的核磁图.溶剂峰以五角星标出
Figure 1.
1H NMR spectra of (A) dibenzylamine, (B) 1b, (C) 2b and (D) polymer P2 in deuterated chloroform (the solvent peaks are marked with asterisks)
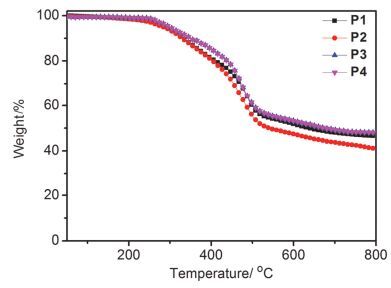 图 2
聚合物的热失重曲线
Figure 2.
TGA curves of polymers
图 2
聚合物的热失重曲线
Figure 2.
TGA curves of polymers
2.2 紫外-可见吸收光谱
聚合物P1~P4的氯仿溶液、固体薄膜及对应单体氯仿溶液的紫外-可见吸收光谱(UV-Vis)见图 3, 具体吸收光谱数据见表 1.如图 3所示, 由于聚合物共轭链被打断, 所有聚合物都呈现尖锐的吸收光谱图, 吸收轮廓基本是对应单体吸收光谱的叠加, 但相比单体的吸收略有红移.此外, 聚合物固态膜的吸收比溶液吸收有不同程度的红移, 说明固态膜下存在聚合物分子链间的聚集.聚合物P1, P2, P3与P4固态薄膜的起始吸收分别为496, 498, 612与620 nm, 因而对应的光学带隙(
${E}_{\text{g}}^{\text{opt}}$ )分别为2.5, 2.49, 2.03与2.00 eV.由于所有的聚合物均为非共轭主链结构, 因此其带隙取决于多组分单体中带隙最窄的单元.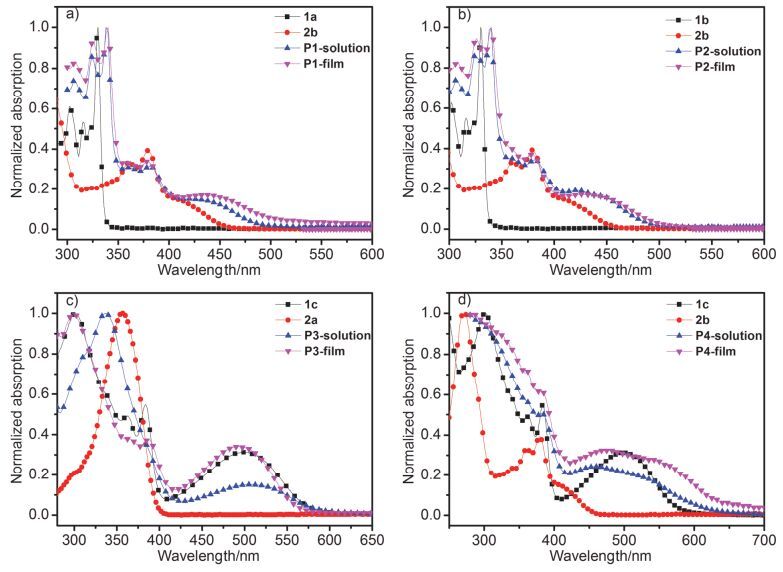 图 3
聚合物的溶液、薄膜以及对应单体的溶液吸收光谱: (a) P1, (b) P2, (c) P3, (d) P4
Figure 3.
The stacked UV-Vis absorption spectra of resultant polymers and their corresponding monomers: (a) P1, (b) P2, (c) P3, (d) P4
图 3
聚合物的溶液、薄膜以及对应单体的溶液吸收光谱: (a) P1, (b) P2, (c) P3, (d) P4
Figure 3.
The stacked UV-Vis absorption spectra of resultant polymers and their corresponding monomers: (a) P1, (b) P2, (c) P3, (d) P4
 表 1
聚合物的光学与电化学数据
Table 1.
UV-Vis absorption and electrochemical properties of polymers
表 1
聚合物的光学与电化学数据
Table 1.
UV-Vis absorption and electrochemical properties of polymers
Polymer $\lambda _{\text{max}}^{\text{solution}}$ /nm$\lambda _{\text{max}}^{\text{film}}$ /nm$\lambda _\text{onset}^\text{film}$ /nm$E_{\text{g}}^{\text{opt}}$ /eV$E_{\text{ox}}^{\text{onset}}$ /V$E_{\text{re}}^{\text{onset}}$ /VEHOMO/eV ELUMO/eV $E_{\text{g}}^{\text{cv}}$/eV P1 339 344 496 2.50 1.64 -0.58 -6.06 -3.84 2.22 P2 339 342 498 2.49 1.68 -0.56 -6.10 -3.86 2.24 P3 338 341 612 2.03 1.48 -0.48 -5.90 -3.94 1.96 P4 — — 620 2.00 1.43 -0.46 -5.85 -3.96 1.89 2.3 电化学性质
通过循环伏安法(CV)研究了聚合物的电化学氧化还原特性.测得的聚合物相对于饱和甘汞的循环伏安曲线见图 4.在相同条件下测得的二茂铁(Fc+/Fc)的氧化还原参比电位为0.39 V, 根据已知二茂铁(Fc+/Fc)对应的真空能级为4.80 eV, 则聚合物的最高占有分子轨道(HOMO)能级与最低未占有分子轨道(LUMO)能级可以由聚合物的起始氧化电位(
$E_{\text{ox}}^{\text{onset}}$ ), 起始还原电位($E_\text{ox}^\text{onset}$ ), 经公式计算得出:${{E}_{\text{HOMO}}}\text{-e(}E_{\text{ox}}^{\text{onset}}\text{+4}\text{.41)eV}$ ;${{E}_{\text{LUMO}}}\text{-e(}E_{\text{re}}^{\text{onset}}\text{+4}\text{.41)eV}$ .计算所得的聚合物具体能级数据见表 1.聚合物P1, P2, P3与P4的$E_{\text{ox}}^{\text{onset}}/E_{\text{re}}^{\text{onset}}$ 分别为1.64/-0.58 V, 1.68/-0.56 V, 1.48/-0.48 V与1.43/-0.46 V, 对应的EHOMO/ELUMO分别为-6.06/-3.84 eV, -6.10/-3.86 eV, -5.90/-3.94 eV与-5.85/-3.96 eV.所有聚合物具有较深的HOMO与LUMO能级, 这是由聚合物骨架给体芴单元与受体NDI单元决定的.聚合物P1与P2表现出相似的能级特征, 因为它们具有完全相同的主链结构. 图 4
聚合物的循环伏安曲线
Figure 4.
CV curves of polymers
图 4
聚合物的循环伏安曲线
Figure 4.
CV curves of polymers
2.4 开尔文探针测试
之前的研究表明带有极性胺基的小分子或者聚合物用作太阳电池阴极界面层时可以有效调节电极的功函, 从而实现活性层与阴极的欧姆接触, 改善电子的抽提[37~39].为了探究主链与侧链胺基不同化学环境对其功函调节的影响, 我们使用开尔文探针测试了聚合物与参比PFN修饰的Ag电极的功函数.测得的未修饰Ag电极的功函数是-4.66 eV, 与文献报道一致[55].当在Ag电极表面旋涂一层聚合物修饰层时, P1与PFN修饰后的Ag电极的功函数分别降至-4.31和-4.27 eV. P2、P3与P4修饰的Ag电极功函数则为-4.61, -4.59与-4.65 eV.尽管P2, P3, P4主链均含有胺基, 却未能有效调节Ag电极的功函.原因可能有两方面:一方面文献报道胺基功能化聚合物侧链富电子氮原子对偶极形成起重要作用[43], 而在聚合物P2, P3, P4中, 主链叔胺的氮由于相连苄基与炔丙基的诱导效应, 氮的电子密度必然大大降低, 因而主链胺基形成偶极的能力大大减弱; 另一方面取代基的位阻效应可能阻碍氮原子孤对电子与金属电极的接触, 进而减弱了主链胺基对金属电极的功函调节能力.
2.5 聚合物电子顺磁共振表征
已有研究表明富电子的胺基可以掺杂缺电子的NDI单元, 进而提高相关NDI聚合物的电荷传输能力[36].为了研究聚合物不同化学环境氮的胺基与NDI掺杂强度之间的关系, 我们对P1与P2在固态下进行了电子顺磁共振(EPR)测试.如图 5所示, 聚合物P1的单电子自旋信号峰是P2信号的15倍, 这表明聚合物P1由胺基向萘并二酰亚胺的电子转移掺杂远远强于P2, 由于P1与P2的主链结构完全一致, 因此, P1表现出更强的自掺杂信号是因为侧链胺基与NDI之间的掺杂.这些结果说明, 氮原子的化学环境对其掺杂能力有较大影响.尽管P2主链含有胺基, 由于与苄基、炔丙基相连, 氮原子的电子密度大大降低, 相应胺基的给电子的能力变弱, 由胺基向NDI单元的电子转移变难, 因而EPR信号较弱.这一现象与前述开尔文探针测试结果一致.
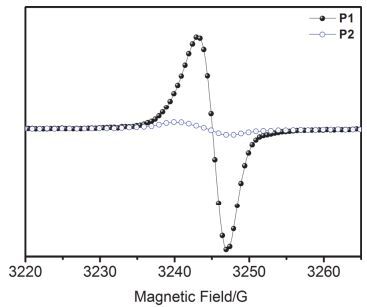 图 5
聚合物P1与P2固态薄膜下的电子顺磁共振谱
Figure 5.
EPR spectra of P1 and P2 in thin films
图 5
聚合物P1与P2固态薄膜下的电子顺磁共振谱
Figure 5.
EPR spectra of P1 and P2 in thin films
2.6 量子化学计算
为了进一步揭示开尔文探针与电子顺磁共振现象的原因并阐明苄基与炔丙基连接的主链胺基与侧链二甲胺基化学环境的本质区别, 我们构建了与P1聚合单元相对应的模型化合物Model S.在B3LYP-D3BJ/ 6-311G (d)[56~58]水平下使用Gaussian 09优化了Model S, 然后通过Multiwfn 3.3.9[59]程序包计算得到原子偶极矩校正的Hirshfeld布居(ADCH)[60]电荷.如图 6所示, 侧链的二甲胺基氮的ADCH电荷为-0.274.而主链胺基氮的ADCH电荷为-0.195, 较侧链胺基氮的负电荷数大大减小.这个结果说明, 由于与苄基、炔丙基相连, 主链胺基上氮的电子密度大大降低.这一结果与前面开尔文探针以及电子顺磁共振表征的结果吻合.
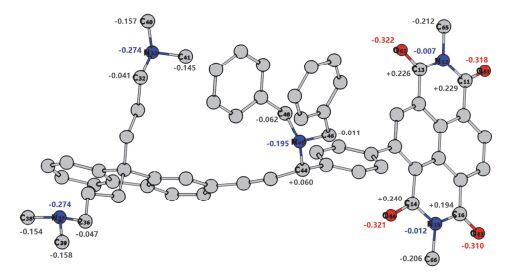 图 6
模型化合物Model S量子化学计算的ADCH电荷
Figure 6.
The ADCH charges of Model S
图 6
模型化合物Model S量子化学计算的ADCH电荷
Figure 6.
The ADCH charges of Model S
2.7 电子迁移率表征
为了研究所得聚合物的电荷传输性质, 我们制备了器件结构为ITO/Al/polymer/Al的单电子器件, 并通过空间电荷限制电流(SCLC)模型拟合数据得到这些聚合物的电子迁移率.单电子器件的电流密度-电压(J-V), 及电流密度1/2-电压(J1/2-V)特征曲线见图 7.通过SCLC曲线的拟合计算, 得到P1, P2, P3与P4的电子迁移率分别为7.2×10-6, 7.7×10-7, 3.3×10-6和6.9×10-6 cm2• V-1•s-1.聚合物表现出相对较低的迁移率可能因为它们的非共轭主链结构.值得一提的是, 尽管P1与P2具有相同的主链结构, P1的电子迁移率比P2高出一个数量级, 这可能是因为P1侧链的胺基与聚合物骨架萘并二酰亚胺具有更强的掺杂效应, 从而有利于提高载流子密度.
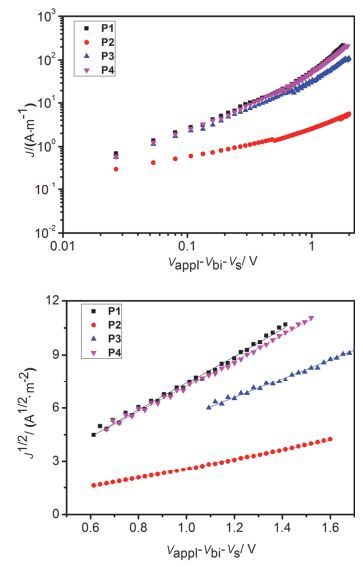 图 7
单电子器件的电流(J, J1/2)-电压(V)特性曲线
Figure 7.
J-V and J1/2-V characteristics of electron-only devices
图 7
单电子器件的电流(J, J1/2)-电压(V)特性曲线
Figure 7.
J-V and J1/2-V characteristics of electron-only devices
2.8 共混薄膜形貌表征
薄膜的形貌对于光电器件的性能有重要影响.通过原子力显微镜(AFM)研究了PTB7-Th:PC71BM共混膜旋涂阴极界面层前后的形貌.如图 8a所示, PTB7-Th:PC71BM共混膜表面较为平整, 表面粗糙度(RMS)值为1.67 nm.当在活性层上再旋涂一层P1后(图 8b), 表面变得更平整, RMS值为1.27 nm, 这说明P1作为阴极界面层能够在一定程度上减小薄膜的粗糙度, 有利于活性层与Al电极的有效接触, 改善电子抽提.
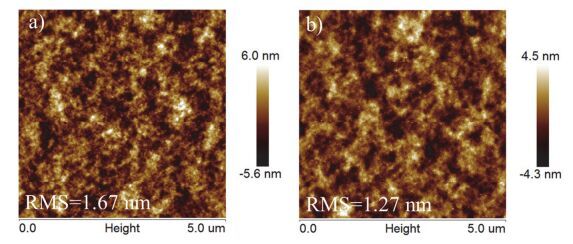 图 8
AFM图: (a) PTB7-Th:PC71BM, (b) PTB7-Th:PC71BM上旋涂P1界面层
Figure 8.
AFM images of PTB7-Th:PC71BM (a) and P1 on the top of PTB7-Th:PC71BM (b)
图 8
AFM图: (a) PTB7-Th:PC71BM, (b) PTB7-Th:PC71BM上旋涂P1界面层
Figure 8.
AFM images of PTB7-Th:PC71BM (a) and P1 on the top of PTB7-Th:PC71BM (b)
2.9 光伏性能
由于P1具有醇溶性, 且能有效的调节高功函金属电极的功函, 因此被用于聚合物太阳电池的阴极界面层.我们制备了器件结构为ITO/PEDOT:PSS/PTB7-Th:PC71BM/P1/Al的聚合物太阳电池器件.活性层化学结构、器件结构图、测试电流(J)-电压(V)曲线与外量子效率(EQE)曲线见图 9, 相应的器件参数见表 2.如图 9所示, 无阴极界面层的参比器件的最大光电转换效率是6.79%, 相应的器件参数短路电流(Jsc)为15.81 mA•cm-2, 开路电压(Voc)为0.67 V, 填充因子(FF)为64.14%.以甲醇处理活性层后, 器件的效率为7.10%.然而, 在活性层与Al电极之间旋涂一层5 nm的P1作为阴极界面层时, 器件的效率提升至9.34% (Jsc=16.23 mA•cm-2, Voc=0.78 V, FF=73.75%).效率的提高是因为开路电压与填充因子的双重提升.开路电压的提升主要是因为P1作为阴极界面层能够在电极表面形成偶极, 有效降低Al电极功函, 因而增加内建电势, 从而获得更高的器件开路电压.此外, 阴极界面层的存在可以改善活性层与电极之间的接触, 改善电子从活性层到阴极的抽提, 显著提升器件的填充因子.为了验证基于电流(J)-电压(V)特性曲线得到的器件的短路电流, 测试了器件的外量子效率曲线(图 9d).结果表明, 基于外量子效率曲线计算的短路电流和采用J-V曲线测试的短路电流值的误差范围<3%, 证实了器件测试结果的可靠性.以上结果表明通过多组分聚合制备的目标聚合物P1可用于聚合物太阳电池阴极界面层, 实现光电转换效率的大幅提升.
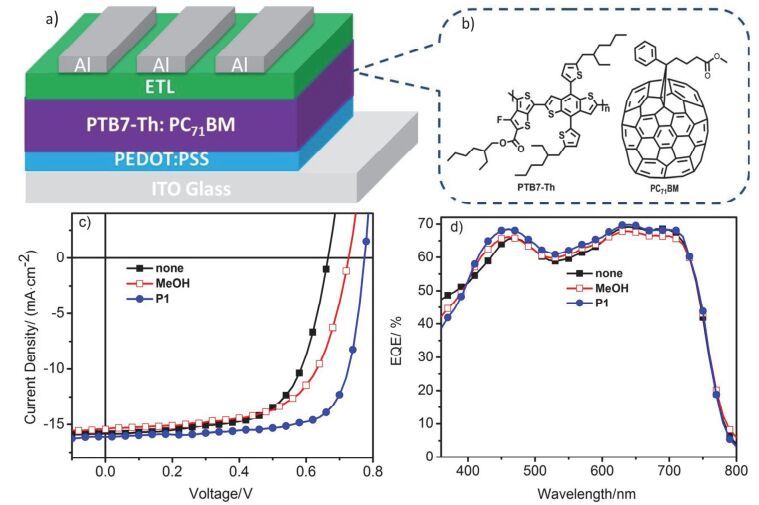 图 9
电池器件结构图(a), 活性层材料的化学结构式(b), 器件的电流(J)-电压(V)特性曲线(c)和外量子效率曲线(d)
Figure 9.
Device configuration of the PSCs (a), the chemical structures of PTB7-Th and PC71BM (b), J-V characteristics (c) and external quantum efficiency spectra (d) of PSCs
表 2
聚合物太阳电池器件参数
Table 2.
Photovoltaic parameters of PSCs
图 9
电池器件结构图(a), 活性层材料的化学结构式(b), 器件的电流(J)-电压(V)特性曲线(c)和外量子效率曲线(d)
Figure 9.
Device configuration of the PSCs (a), the chemical structures of PTB7-Th and PC71BM (b), J-V characteristics (c) and external quantum efficiency spectra (d) of PSCs
表 2
聚合物太阳电池器件参数
Table 2.
Photovoltaic parameters of PSCs
Cathode interlayer Jsc/(mA•cm-2) Voc/V FF/% PCEmax/% none 15.81 0.67 64.14 6.79 MeOH 15.44 0.73 63.02 7.10 P1 16.23 0.78 73.75 9.34 3 结论
基于炔、醛、胺三种单体, 通过微波辅助一锅法三组分聚合合成了一系列胺基功能化聚合物, 并研究了不同化学环境的侧链与主链胺基对功函调节、自掺杂行为的影响.开尔文探针与电子顺磁共振谱研究表明, 胺基调节电极功函、掺杂NDI的功能与胺基氮的化学环境有关.当胺基上取代基团导致氮的电子云密度降低, 位阻增大时, 胺基形成界面偶极, 进而调节电极功函的能力减弱或消失.此外, 胺基氮的电子云密度降低还会导致富电子的胺基向缺电子单元NDI的电子转移掺杂的强度大大减弱.在P1~P4中, P1因具有良好的醇溶性, 可被用于聚合物太阳电池的阴极界面层.以PTB7-Th:PC71BM为活性层, P1为阴极界面层制备的聚合物太阳电池器件光电转换效率达到9.34%.
4 实验部分
4.1 试剂与仪器
石油醚、二氯甲烷、甲醇等试剂购自广州化学试剂厂, 如无特别说明则直接使用.四氢呋喃用金属钠处理, 以二苯甲酮作指示剂蒸馏得到.多组分反应在Discover SP微波合成仪上进行. 1H NMR谱和13C NMR谱使用BrukerAV500核磁共振谱仪分别在500和125 MHz常温下测得, CDCl3为溶剂, TMS为内标.聚合物分子量在Waters GPC 2410凝胶渗透色谱(GPC)上测得, 流动相为THF, 使用标准聚苯乙烯校准曲线.热重分析曲线在NETZSCH TG209热重分析仪上测得, 升温速率为10 K•min-1. UV-vis吸收光谱使用HP 8453紫外-可见光光谱仪测得.聚合物的氧化还原行为用循环伏安法在CHI 660D电化学工作站上采集.测试采用标准的三电极即玻碳电极为工作电极, 铂丝电极为对电极, 甘汞电极(SCE)为参比电极, 以二茂铁作为标准物校准, 在0.1 mol/L四丁基六氟磷酸铵(TBAPF6)的乙腈溶液中测得.形貌表征使用DINS4 AFM (Veeco)原子力显微镜在Tapping模式下测得.
4.2 电池器件的制备
ITO导电玻璃依次用去离子水, 丙酮, 异丙醇超声洗涤后放入真空烘箱干燥过夜.干燥后的ITO用O2-plasma处理5 min.随后2500 r•min-1旋涂一层40 nm厚PEDOT:PSS (Baytron P4083, Bayer AG), 150 ℃热处理20 min.将聚合物PTB7-Th:PC71BM (1:1.5) 的氯苯溶液旋涂在PEDOT:PSS空穴传输层上, 得到约100 nm厚的活性层.将聚合物P1配成1 mg•mL-1的甲醇溶液, 旋涂在活性层上, 获得约5 nm左右的阴极界面层.将器件放入2×10-6 mbar压力下的高真空蒸镀仓, 通过真空热蒸镀在阴极界面表面蒸镀上一层100 nm的Al电极.器件的有效面积通过掩模板调控为0.04 cm2, 同时蒸镀完Al阴极的器件用环氧树脂紫外光固化包封.能量转换效率在1个模拟太阳光下获得(Japan, SAN-EI, XES-40S1).太阳光模拟器采用标准的硅电池标定为100 mW•cm-2. J-V特征曲线采用Keithley 2400采集.所有的器件制备过程都在氮气填充的手套箱中完成(Braun GmbH).
4.3 单体与聚合物的制备
化合物5[36]、8[61]、1a[49]、1b[49]、2a[49]根据之前报道的文献合成, 具体合成路线如图 1所示.
2, 6-(双-5-乙炔基-2-噻吩基)-N, N'-二异辛基-1, 4, 5, 8-萘并二酰亚胺(1c):将化合物5 (4.0 g, 5.00 mmol), CuI (0.026 g, 0.14 mmol), 二三苯基膦二氯化钯(0.175 g, 0.25 mmol)加入两口圆底烧瓶.反应体系抽换气三次并填充氩气, 使用注射器加入甲苯(100 mL)与二异丙基胺(30 mL).随后将三甲基乙炔基硅(3 mL)以15 mL二异丙胺稀释后逐滴滴加至反应瓶, 反应液70 ℃搅拌4 h.待反应液冷却至室温, 二氯甲烷萃取三次, 有机相减压旋蒸除去溶剂, 粗产物以二氯甲烷为淋洗剂硅胶柱层析得到红色固体6.化合物6直接溶于100 mL二氯甲烷/甲醇(V:V=1:1) 混合液, 加入K2CO3 (1 g)搅拌1 h.反应液倒入水(200 mL)中, 二氯甲烷萃取, 有机相减压旋蒸除去溶剂后, 粗产物以石油醚/二氯甲烷(V:V=1:1) 硅胶柱层析得到紫红色固体1c (2.47 g, 两步合计产率72%).1H NMR (500 MHz, CDCl3) δ: 8.72 (s, 2H), 7.34 (d, J=3.8 Hz, 2H), 7.15 (d, J=3.7 Hz, 2H), 4.07 (dd, J=7.4, 4.5 Hz, 4H), 3.48 (s, 2H), 1.89 (dt, J=12.9, 6.5 Hz, 2H), 1.36~1.26 (m, 16H), 0.89 (dt, J=14.1, 7.3 Hz, 12H). 13C NMR (125 MHz, CDCl3) δ: 162.5, 162.2, 142.4, 136.5, 133.3, 129.9, 128.1, 127.6, 125.6, 124.9, 123.6, 83.2, 76.5, 44.7, 37.7, 30.6, 28.5, 23.9, 23.1, 14.1, 10.6.
2, 6-(双-4-甲酰基-1-苯基)-N, N'-二异辛基-1, 4, 5, 8-萘并二酰亚胺(2b):将化合物8 (4.0 g, 5.00 mmol), 4-甲酰基苯硼酸(1.88 g, 12.5 mmol), 四三苯基膦钯(0.289 g, 0.25 mmol), 加入到两口圆底烧瓶.反应体系抽换气三次并填充氩气, 随后使用注射器加入脱气的K2CO3水溶液(10 mL 2 mol•L-1)与甲苯(100 mL).反应在100 ℃回流搅拌反应12 h, 冷却至室温, 水洗, 二氯甲烷萃取三次, 合并有机相, 减压旋蒸除去溶剂后, 粗产物以石油醚/二氯甲烷(V:V=1:1) 硅胶柱层析得到黄色粉末2b. 1H NMR (500 MHz, CDCl3) δ: 10.14 (s, 2H), 8.65 (s, 2H), 8.05 (d, J=8.1 Hz, 4H), 7.57 (d, J=8.1 Hz, 4H), 4.03 (dd, J=7.4, 4.4 Hz, 4H), 1.86 (dt, J=12.8, 6.4 Hz, 2H), 1.33~1.22 (m, 16H), 0.87 (dt, J=11.7, 7.2 Hz, 12H); 13C NMR (125 MHz, CDCl3) δ: 191.7, 162.5, 162.4, 146.6, 146.4, 135.9, 135.2, 129.8, 128.8, 127.4, 125.9, 123.2, 44.6, 37.7, 30.5, 28.5, 23.8, 23.1, 14.1, 10.6.
P1的合成:将单体1a (115.2 mg, 0.3 mmol), 2b (209.4 mg, 0.3 mmol), 二苄胺(130 mg, 0.66 mmol)加入到35 mL的微波反应管.反应管在手套箱过渡仓抽换气三次除去氧气.之后在手套箱中加入InCl3(13.2 mg, 0.06 mmol), 4 Å分子筛(100 mg)、邻二甲苯(3.0 mL).反应装置移出手套箱, 在微波反应器中160 ℃反应45 min.待反应液冷却至室温, 经0.45 μm PTFE滤头过滤, 逐滴滴加沉淀在甲醇(150 mL)中.收集固体再次溶解沉淀, 所得产物40 ℃干燥至恒重, 得棕色固体P1, Mw=12300; Mw/Mn=1.31. 1H NMR (500 MHz, CDCl3) δ: 8.65 (s, 2H), 8.06~7.92 (m, 4H), 7.77~7.64 (m, 6H), 7.51 (m, 8H), 7.43~7.36 (m, 20H), 5.12 (s, 2H), 4.06~4.01 (m, 4H), 3.93 (d, J =12.6 Hz, 4H), 3.68 (d, J =12.2 Hz, 4H), 2.09~1.86 (m, 22H), 1.36~1.18 (m, 16H), 0.86~0.84 (m, 16H); 13C NMR (125 MHz, CDCl3) δ: 162.9, 162.6, 150.6, 147.5, 140.7, 140.2, 139.5, 136.0, 131.5, 129.2, 129.1, 128.4, 127.1, 126.2, 125.5, 122.9, 120.9, 120.1, 89.6, 85.0, 59.7, 56.2, 55.0, 54.9, 45.4, 37.6, 30.5, 28.4, 23.9, 23.1, 22.1, 14.1, 10.6.
P2的合成:相同的操作, 单体1b (97.8 mg, 0.3 mmol), 2b (209.4 mg, 0.3 mmol), 二苄胺(130 mg, 0.66 mmol), InCl3 (13.2 mg, 0.06 mmol), 4 Å分子筛(100 mg), 邻二甲苯(3.0 mL), 150 ℃微波聚合反应45min得棕色产物P2 (339 mg, 产率82%), Mw=20800; Mw/Mn=1.52.1H NMR (500 MHz, CDCl3) δ: 8.65 (s, 2H), 8.06~7.91 (m, 4H), 7.78~7.65 (m, 6H), 7.52~7.50 (m, 8H), 7.43~7.33 (m, 20H), 5.12 (s, 2H), 4.05~4.00 (m, 4H), 3.93 (d, J =13.2 Hz, 4H), 3.68 (d, J =12.8 Hz, 4H), 2.09~1.85 (m, 6H), 1.30~1.25 (m, 20H), 0.85~0.72 (m, 22H); 13C NMR (125 MHz, CDCl3) δ: 162.9, 162.6, 151.3, 147.5, 140.7, 139.5, 135.9, 131.3, 129.2, 129.0, 128.9, 128.4, 128.0, 127.1, 126.2, 125.5, 123.0, 120.9, 120.0, 89.7, 84.9, 56.2, 55.3, 54.9, 44.4, 40.1, 37.7, 30.5, 28.4, 26.0, 23.9, 23.1, 14.1, 10.6.
P3的合成:相同的操作, 单体1c (206.7 mg, 0.3 mmol), 单体2a (145.8 mg, 0.3 mmol), 二苄胺(130 mg, 0.66 mmol), InCl3 (13.2 mg, 0.06 mmol), 4 Å分子筛(100 mg), 邻二甲苯(3.0 mL), 140 ℃微波聚合反应30 min得紫色固体P3 (358 mg, yield 78%). Mw=22600, Mw/Mn=1.61. 1H NMR (500 MHz, CDCl3) δ: 8.84 (s, 2H), 8.02~8.01 (m, 4H), 7.83 (t, J=10.9 Hz, 6H), 7.72~7.58 (m, 4H), 7.51~7.33 (m, 22H), 7.18~7.14 (m, 2H), 5.07 (s, 2H), 4.15~4.09 (m, 4H), 3.95 (d, J=13.4 Hz, 4H), 3.65 (d, J=13.5 Hz, 4H), 2.06~1.96 (m, 6H), 1.43~1.31 (m, 16H), 1.13~1.10 (m, 4H), 0.98~0.89 (m, 12H), 0.72~0.68 (m, 10H); 13C NMR (125 MHz, CDCl3) δ: 162.5, 162.4, 151.7, 147.7, 142.4, 141.7, 141.0, 140.1, 139.4, 136.6, 135.0, 133.3, 132.4, 130.3, 129.2, 128.9, 128.7, 128.4, 127.7, 127.4, 127.1, 127.0, 126.2, 126.0, 125.6, 123.4, 121.5, 120.9, 91.6, 81.4, 56.3, 55.3, 55.2, 44.7, 40.3, 37.8, 30.7, 28.6, 26.0, 24.0, 23.1, 22.1, 14.1, 13.8, 10.7.
P4的合成:相同的操作, 单体1c (206.7 mg, 0.3 mmol), 2b (209.4 mg, 0.3 mmol), 二苄胺(130 mg, 0.66 mmol), InCl3(13.2 mg, 0.06 mmol), 4 Å分子筛(100 mg), 邻二甲苯(3.0 mL), 140 ℃微波聚合反应30 min得紫色固体P4 (361 mg, 产率69%), Mw=26600; Mw/Mn=1.80. 1H NMR (500 MHz, CDCl3) δ: 8.83 (s, 2H), 8.65 (s, 2H), 7.86 (d, J=6.8 Hz, 4H), 7.50~7.35 (m, 22H), 7.29~7.25 (m, 4H), 7.17~7.15 (m, 2H), 5.12 (s, 2H), 4.13~4.01 (m, 8H), 3.92 (d, J=13.4 Hz, 4H), 3.61 (d, J=13.4 Hz, 4H), 2.01~1.85 (m, 4H), 1.41~1.25 (m, 32H), 0.92~0.83 (m, 24H); 13C NMR (125 MHz, CDCl3) δ: 162.9, 162.6, 162.5, 162.4, 146.7, 142.4, 141.8, 139.3, 136.6, 135.9, 135.3, 133.3, 129.9, 129.2, 129.0, 128.9, 128.4, 128.1, 127.7, 127.2, 125.5, 123.5, 123.0, 120.9, 91.4, 81.5, 56.4, 54.9, 44.7, 44.5, 37.6, 30.6, 29.7, 29.3, 28.6, 28.5, 24.0, 23.9, 23.1, 14.1, 10.6.
-
-
[1]
Domling, A.; Ugi, I. Angew. Chem., Int. Ed. 2000, 39, 3168. doi: 10.1002/(ISSN)1521-3773
-
[2]
Kakuchi, R. Angew. Chem., Int. Ed. 2014, 53, 46. doi: 10.1002/anie.v53.1
-
[3]
Balme, G.; Bossharth, E.; Monteiro, N. Eur. J. Org. Chem. 2003, 2003, 4101. doi: 10.1002/(ISSN)1099-0690
-
[4]
Andreana, P. R.; Liu, C. C.; Schreiber, S. L. Org. Lett. 2004, 6, 4231. doi: 10.1021/ol0482893
-
[5]
D'Souza, D. M.; Mueller, T. J. J. Chem. Soc. Rev. 2007, 36, 1095. doi: 10.1039/B608235C
-
[6]
Biggs-Houck, J. E.; Younai, A.; Shaw, J. T. Curr. Opin. Chem. Biol. 2010, 14, 371. doi: 10.1016/j.cbpa.2010.03.003
-
[7]
Siamaki, A. R.; Sakalauskas, M.; Arndtsen, B. A. Angew. Chem., Int. Ed. 2011, 50, 6552. doi: 10.1002/anie.v50.29
-
[8]
Thanh Binh, N.; Minh Quan, T.; Ermolenko, L.; Al-Mourabit, A. Org. Lett. 2014, 16, 310. doi: 10.1021/ol403345e
-
[9]
Rotstein, B. H.; Zaretsky, S.; Rai, V.; Yudin, A. K. Chem. Rev. 2014, 114, 8323. doi: 10.1021/cr400615v
-
[10]
Levi, L.; Muller, T. J. J. Chem. Soc. Rev. 2016, 45, 2825. doi: 10.1039/C5CS00805K
-
[11]
Teimouri, M. B.; Abbasi, T.; Mivehchi, H. Tetrahedron 2008, 64, 10425. doi: 10.1016/j.tet.2008.08.039
-
[12]
Yu, J.; Shi, F.; Gong, L.-Z. Acc. Chem. Res. 2011, 44, 1156. doi: 10.1021/ar2000343
-
[13]
Ruijter, E.; Scheffelaar, R.; Orru, R. V. A. Angew. Chem., Int. Ed. 2011, 50, 6234. doi: 10.1002/anie.201006515
-
[14]
Hossaini, Z.; Seyfi, S.; Rostami-Charati, F.; Ghambarian, M. Comb. Chem. High Throughput Screen 2013, 16, 788. doi: 10.2174/13862073113169990046
-
[15]
Pagadala, R.; Kommidi, D. R.; Kankala, S.; Maddila, S.; Singh, P.; Moodley, B.; Koorbanally, N. A.; Jonnalagadda, S. B. Org. Biomol. Chem. 2015, 13, 1800. doi: 10.1039/C4OB02229G
-
[16]
Theato, P., Multi-Component and Sequential Reactions in Polymer Synthesis, Springer, 2015, Vol. 269.
-
[17]
Leitch, D. C.; Kayser, L. V.; Han, Z.-Y.; Siamaki, A. R.; Keyzer, E. N.; Gefen, A.; Arndtsen, B. A. Nature Commun. 2015, 6, 7411. doi: 10.1038/ncomms8411
-
[18]
Hu, R. R.; Li, W. Z.; Tang, B. Z. Macromol. Chem. Phys. 2016, 217, 213. doi: 10.1002/macp.201500291
-
[19]
Kreye, O.; Toth, T.; Meier, M. A. R. J. Am. Chem. Soc. 2011, 133, 1790. doi: 10.1021/ja1113003
-
[20]
Lee, I.-H.; Kim, H.; Choi, T.-L. J. Am. Chem. Soc. 2013, 135, 3760. doi: 10.1021/ja312592e
-
[21]
Chan, C. Y. K.; Tseng, N.-W.; Lam, J. W. Y.; Liu, J. Z.; Kwok, R. T. K.; Tang, B. Z. Macromolecules 2013, 46, 3246. doi: 10.1021/ma4005346
-
[22]
Liu, Y. J.; Gao, M.; Lam, J. W. Y.; Hu, R. R.; Tang, B. Z. Macromolecules 2014, 47, 4908. doi: 10.1021/ma501477w
-
[23]
Deng, H. Q.; Hu, R. R.; Zhao, E. G.; Chan, C. Y. K.; Lam, J. W. Y.; Tang, B. Z. Macromolecules 2014, 47, 4920. doi: 10.1021/ma501190g
-
[24]
Li, W. Z.; Wu, X. Y.; Zhao, Z. J.; Qin, A. J.; Hu, R. R.; Tang, B. Z. Macromolecules 2015, 48, 7747. doi: 10.1021/acs.macromol.5b02193
-
[25]
Zheng, C.; Deng, H. Q.; Zhao, Z. J.; Qin, A. J.; Hu, R. R.; Tang, B. Z. Macromolecules 2015, 48, 1941. doi: 10.1021/acs.macromol.5b00175
-
[26]
Deng, H. Q.; Hu, R. R.; Leung, A. C. S.; Zhao, E. G.; Lam, J. W. Y.; Tang, B. Z. Polym. Chem. 2015, 6, 4436. doi: 10.1039/C5PY00477B
-
[27]
Yu, G.; Gao, J.; Hummelen, J. C.; Wudl, F.; Heeger, A. J. Science 1995, 270, 1789. doi: 10.1126/science.270.5243.1789
-
[28]
Heeger, A. J. Chem. Soc. Rev. 2010, 39, 2354. doi: 10.1039/b914956m
-
[29]
Hains, A. W.; Liang, Z.; Woodhouse, M. A.; Gregg, B. A. Chem. Rev. 2010, 110, 6689. doi: 10.1021/cr9002984
-
[30]
Huang, Y.; Kramer, E. J.; Heeger, A. J.; Bazan, G. C. Chem. Rev. 2014, 114, 7006. doi: 10.1021/cr400353v
-
[31]
Lu, L. Y.; Zheng, T. Y.; Wu, Q. H.; Schneider, A. M.; Zhao, D. L.; Yu, L. P. Chem. Rev. 2015, 115, 12666. doi: 10.1021/acs.chemrev.5b00098
-
[32]
Zhang, X.; Wang, Z. L.; Chen, S. Y.; Zhao, Z.; Yuan, W.; Wang, H. P.; Gao, X. K. Chin. J. Chem. 2014, 32, 1057.
-
[33]
赵蔡斌, 王占领, 周科, 葛红光, 张强, 靳玲侠, 王文亮, 尹世伟, 化学学报, 2015, 74, 251. doi: 10.3969/j.issn.0253-2409.2015.02.017Zhao, C. B.; Wang, Z. L.; Zhou, K.; Ge, H. G.; Zhang, Q.; Jin, L. X.; Wang, W. L.; Yin, S. W. Acta Chim. Sinica 2015, 74, 251. doi: 10.3969/j.issn.0253-2409.2015.02.017
-
[34]
Liu, L. Q.; Zhang, G. C.; He, B. T.; Huang, F. Chin. J. Chem. 2015, 33, 902.
-
[35]
Zhang, Z.-G.; Qi, B.; Jin, Z.; Chi, D.; Qi, Z.; Li, Y.; Wang, J. Energy Environ. Sci. 2014, 7, 1966. doi: 10.1039/c4ee00022f
-
[36]
Wu, Z. H.; Sun, C.; Dong, S.; Jiang, X.-F.; Wu, S. P.; Wu, H. B.; Yip, H.-L.; Huang, F.; Cao, Y. J. Am. Chem. Soc. 2016, 138, 2004. doi: 10.1021/jacs.5b12664
-
[37]
张凯, 管星, 黄飞, 曹镛, 化学学报, 2012, 70, 2489. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract341751.shtmlZhang, K.; Guan, X.; Huang, F.; Cao, Y. Acta Chim. Sinica 2012, 70, 2489. http://sioc-journal.cn/Jwk_hxxb/CN/abstract/abstract341751.shtml
-
[38]
Duan, C. H.; Zhang, K.; Zhong, C. M.; Huang, F.; Cao, Y. Chem. Soc. Rev. 2013, 42, 9071. doi: 10.1039/c3cs60200a
-
[39]
Hu, Z. C.; Zhang, K.; Huang, F.; Cao, Y. Chem. Commun. 2015, 51, 5572. doi: 10.1039/C4CC09433F
-
[40]
Yip, H.-L.; Jen, A. K. Y. Energy Environ. Sci. 2012, 5, 5994. doi: 10.1039/c2ee02806a
-
[41]
卢俊明, 蔡万清, 张桂传, 刘升建, 应磊, 黄飞, 化学学报, 2015, 73, 1153. doi: 10.3866/PKU.WHXB201504145Lu, J. M.; Cai, W. Q.; Zhang, G. C.; Liu, S. J.; Ying, L.; Huang, F. Acta Chim. Sinica 2015, 73, 1153. doi: 10.3866/PKU.WHXB201504145
-
[42]
Zhang, K.; Hu, Z. C.; Sun, C.; Wu, Z. H.; Huang, F.; Cao, Y. Chem. Mater. 2017, 29, 141. doi: 10.1021/acs.chemmater.6b02802
-
[43]
van Reenen, S.; Kouijzer, S.; Janssen, R. A. J.; Wienk, M. M.; Kemerink, M. Adv. Mater. Inter. 2014, 1, 1400189. doi: 10.1002/admi.201400189
-
[44]
Hu, Z. H.; Zhong, Z. M.; Chen, Y. W.; Sun, C.; Huang, F.; Peng, J. B.; Wang, J.; Cao, Y. Adv. Funct. Mater. 2016, 26, 129. doi: 10.1002/adfm.201503420
-
[45]
Liu, S. J.; Zhang, K.; Lu, J. M.; Zhang, J.; Yip, H. L.; Huang, F.; Cao, Y. J. Am. Chem. Soc. 2013, 135, 15326. doi: 10.1021/ja408363c
-
[46]
Liu, X.; Xu, R. G.; Duan, C.; Huang, F.; Cao, Y. J. Mater. Chem. C 2016, 4, 4288.
-
[47]
Guan, X.; Zhang, K.; Huang, F.; Bazan, G. C.; Cao, Y. Adv. Funct. Mater. 2012, 22, 2846. doi: 10.1002/adfm.v22.13
-
[48]
Kan, Y. Y.; Zhu, Y. X.; Liu, Z. L.; Zhang, L. J.; Chen, J. W.; Cao, Y. Macromol. Rapid Commun. 2015, 36, 1393. doi: 10.1002/marc.v36.15
-
[49]
Jia, T.; Zheng, N. N.; Cai, W. Q.; Zhang, J.; Ying, L.; Huang, F.; Cao, Y. Chin. J. Polym. Sci. 2017, 35, 269.
-
[50]
Yan, H.; Chen, Z. H.; Zheng, Y.; Newman, C.; Quinn, J. R.; Dötz, F.; Kastler, M.; Facchetti, A. Nature 2009, 457, 679. doi: 10.1038/nature07727
-
[51]
Bucella, S. G.; Luzio, A.; Gann, E.; Thomsen, L.; McNeill, C. R.; Pace, G.; Perinot, A.; Chen, Z. H.; Facchetti, A.; Caironi, M. Nature Commun. 2015, 6, 8394. doi: 10.1038/ncomms9394
-
[52]
Facchetti, A. Mater. Today 2013, 16, 123. doi: 10.1016/j.mattod.2013.04.005
-
[53]
Mu, C.; Liu, P.; Ma, W.; Jiang, K.; Zhao, J.; Zhang, K.; Chen, Z. H.; Wei, Z. H.; Yi, Y.; Wang, J. N.; Yang, S. H.; Huang, F.; Facchetti, A.; Ade, H.; Yan, H. Adv. Mater. 2014, 26, 7224. doi: 10.1002/adma.v26.42
-
[54]
Fabiano, S.; Himmelberger, S.; Drees, M.; Chen, Z.; Altamimi, R. M.; Salleo, A.; Loi, M. A.; Facchetti, A. Adv. Energy Mater. 2014, 4, 1301409. doi: 10.1002/aenm.201301409
-
[55]
Sun, C.; Wu, Z. H.; Yip, H.-L.; Zhang, H.; Jiang, X.-F.; Xue, Q.; Hu, Z. C.; Hu, Z.; Shen, Y.; Wang, M.; Huang, F.; Cao, Y. Adv. Energy Mater. 2016, 6, 15011534.
-
[56]
Krishnan, R.; Binkley, J. S.; Seeger, R.; Pople, J. A. J. Chem. Phys. 1980, 72, 650.
-
[57]
McLean, A.; Chandler, G. J. Chem. Phys. 1980, 72, 5639.
-
[58]
Becke, A. D. J. Chem. Phys. 1993, 98, 5648.
-
[59]
Lu, T.; Chen, F. W. J. Comput. Chem. 2012, 33, 580. doi: 10.1002/jcc.v33.5
-
[60]
Lu, T.; Chen, F. W. J. Theor. Comput. Chem. 2012, 11, 163. doi: 10.1142/S0219633612500113
-
[61]
Bhosale, S. V.; Jani, C. H.; Langford, S. J. Chem. Soc. Rev. 2008, 37, 331. doi: 10.1039/B615857A
-
[1]
-

图 1 (A)二苄胺, (B) 1b, (C) 2b, 聚合物P2在氘代氯仿的核磁图.溶剂峰以五角星标出
Figure 1 1H NMR spectra of (A) dibenzylamine, (B) 1b, (C) 2b and (D) polymer P2 in deuterated chloroform (the solvent peaks are marked with asterisks)

图 3 聚合物的溶液、薄膜以及对应单体的溶液吸收光谱: (a) P1, (b) P2, (c) P3, (d) P4
Figure 3 The stacked UV-Vis absorption spectra of resultant polymers and their corresponding monomers: (a) P1, (b) P2, (c) P3, (d) P4

图 7 单电子器件的电流(J, J1/2)-电压(V)特性曲线
Figure 7 J-V and J1/2-V characteristics of electron-only devices

图 8 AFM图: (a) PTB7-Th:PC71BM, (b) PTB7-Th:PC71BM上旋涂P1界面层
Figure 8 AFM images of PTB7-Th:PC71BM (a) and P1 on the top of PTB7-Th:PC71BM (b)

图 9 电池器件结构图(a), 活性层材料的化学结构式(b), 器件的电流(J)-电压(V)特性曲线(c)和外量子效率曲线(d)
Figure 9 Device configuration of the PSCs (a), the chemical structures of PTB7-Th and PC71BM (b), J-V characteristics (c) and external quantum efficiency spectra (d) of PSCs
表 1 聚合物的光学与电化学数据
Table 1. UV-Vis absorption and electrochemical properties of polymers
Polymer $\lambda _{\text{max}}^{\text{solution}}$ /nm$\lambda _{\text{max}}^{\text{film}}$ /nm$\lambda _\text{onset}^\text{film}$ /nm$E_{\text{g}}^{\text{opt}}$ /eV$E_{\text{ox}}^{\text{onset}}$ /V$E_{\text{re}}^{\text{onset}}$ /VEHOMO/eV ELUMO/eV $E_{\text{g}}^{\text{cv}}$/eV P1 339 344 496 2.50 1.64 -0.58 -6.06 -3.84 2.22 P2 339 342 498 2.49 1.68 -0.56 -6.10 -3.86 2.24 P3 338 341 612 2.03 1.48 -0.48 -5.90 -3.94 1.96 P4 — — 620 2.00 1.43 -0.46 -5.85 -3.96 1.89  下载: 导出CSV
下载: 导出CSV
表 2 聚合物太阳电池器件参数
Table 2. Photovoltaic parameters of PSCs
Cathode interlayer Jsc/(mA•cm-2) Voc/V FF/% PCEmax/% none 15.81 0.67 64.14 6.79 MeOH 15.44 0.73 63.02 7.10 P1 16.23 0.78 73.75 9.34
下载: 导出CSV
-











 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 12
- 文章访问数: 2406
- HTML全文浏览量: 477
 下载:
下载:
