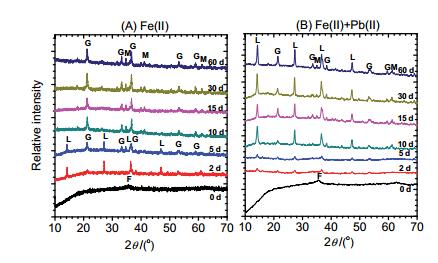
 图 1
水铁矿晶相转变过程中的XRD图谱: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系(F:水铁矿; L:纤铁矿; G:针铁矿; M:磁铁矿)
Figure 1.
XRD patterns of the transformed ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)(F: ferrihydrite; L: lepidocrocite; G: Goethite; M: magnetite)
图 1
水铁矿晶相转变过程中的XRD图谱: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系(F:水铁矿; L:纤铁矿; G:针铁矿; M:磁铁矿)
Figure 1.
XRD patterns of the transformed ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)(F: ferrihydrite; L: lepidocrocite; G: Goethite; M: magnetite)
引用本文:
刘承帅, 李芳柏, 陈曼佳, 廖长忠, 童辉, 华健. Fe(Ⅱ)催化水铁矿晶相转变过程中Pb的吸附与固定[J]. 化学学报,
2017, 75(6): 621-628.
doi:
10.6023/A17030093

Citation: Liu Chengshuai, Li Fangbai, Chen Manjia, Liao Changzhong, Tong Hui, Hua Jian. Adsorption and Stabilization of Lead during Fe(Ⅱ)-catalyzed Phase Transformation of Ferrihydrite[J]. Acta Chimica Sinica, 2017, 75(6): 621-628. doi: 10.6023/A17030093
Citation: Liu Chengshuai, Li Fangbai, Chen Manjia, Liao Changzhong, Tong Hui, Hua Jian. Adsorption and Stabilization of Lead during Fe(Ⅱ)-catalyzed Phase Transformation of Ferrihydrite[J]. Acta Chimica Sinica, 2017, 75(6): 621-628. doi: 10.6023/A17030093
Fe(Ⅱ)催化水铁矿晶相转变过程中Pb的吸附与固定
摘要:
厌氧状态下,游离态Fe(Ⅱ)(Fe(Ⅱ)aq)催化氧化铁晶相重组是重要的铁循环化学过程,其本质是Fe(Ⅱ)aq与结构态Fe(Ⅲ)间的Fe原子交换,这一过程对稻田土壤和沉积物中重金属的环境行为产生重要影响,其影响机制有待于深入研究.本研究结果显示,Fe(Ⅱ)aq催化水铁矿晶相转变过程中,重金属离子Pb(Ⅱ)通过与Fe(Ⅱ)的竞争性吸附,降低了水铁矿表面吸附态Fe(Ⅱ)浓度,抑制了Fe(Ⅱ)aq与水铁矿中结构态Fe(Ⅲ)之间的Fe原子交换,最终降低水铁矿晶相转变速率并改变水铁矿晶相转变途径.无Pb(Ⅱ)时,水铁矿最终转变为针铁矿和磁铁矿;Pb(Ⅱ)影响下,转变产物主要为纤铁矿,部分为针铁矿和磁铁矿.在水铁矿晶相转变过程中,部分吸附到氧化铁表面的Pb(Ⅱ)通过晶体包裹或Fe结构位取代,被形成的氧化铁结构化固定,从而降低了重金属Pb(Ⅱ)的活性.
English
Adsorption and Stabilization of Lead during Fe(Ⅱ)-catalyzed Phase Transformation of Ferrihydrite
Abstract:
Aqueous Fe(Ⅱ) (Fe(Ⅱ)aq)-catalyzed recrystallization of iron (hydr)oxides is the important chemical reaction of iron cycle in anaerobic environments, which poses significant effects on the environmental behavior of heavy metals in soils and sediments. Ferrihydrite is the initial iron mineral phase during the ferrous mineralization and has relatively unstable crystal structure. The structure transformation behavior of ferrihydrite is active and also poses important effects on environmental behavior of soil heavy metals. However, the Fe(Ⅱ)aq-catalyzed phase transformation of ferrihydrite has been rarely reported, especially with the coexisting metal ions. In the present study, the effects of coexisting heavy metal of Pb(Ⅱ) on the Fe(Ⅱ)aq-catalyzed phase transformation of ferrihydrite coupling the environmental behavior of Pb(Ⅱ) were systematically studied. The results show that ferrihydrite underwent efficient phase transformation rates when catalyzed by Fe(Ⅱ)aq whenever with or without the effect of Pb(Ⅱ). Compared with the reaction system that without Pb(Ⅱ), the adsorption of Fe(Ⅱ) on the surface of ferrihydrite was inhibited due to the competition of Pb(Ⅱ) when with the coexistence of Pb(Ⅱ), which further decreased the rates of Fe atom exchange between Fe(Ⅱ)aq and structural Fe(Ⅲ) of ferrihydrite. With the inhibited Fe atom exchange reaction, the phase transformation rates were relatively decreased and transformation products were changed during the Fe(Ⅱ)aq-catalyzed phase transformation of ferrihydrite. Goethite and magnetite were found to be the final transformed products of iron (hydr)oxides when without Pb(Ⅱ), while lepidocrocite was determined to be the main transformed product with little goethite and magnetite as the other transformed products when with Pb(Ⅱ). During the Fe(Ⅱ)aq-catalyzed phase transformation of ferrihydrite with the coexistence of Pb(Ⅱ), some Pb were stabilized through being incorporated into the structure of ferrihydrite transformed products with the possible mechanisms of occlusion by the crystal lattice and structural incorporation, so as to decrease the activity of the polluted heavy metal of Pb. The obtained results in the present study are expected to provide further insights for understanding the iron cycle coupling with the environmental behavior of heavy metals in soils and sediments.
-
Key words:
- iron oxide
- / Fe atom exchange
- / heavy metal
- / stable isotope tracing
- / isomorphous replacement
-
1 引言
土壤游离态Fe(Ⅱ)(Fe(Ⅱ)aq)催化氧化铁晶相重组是铁循环的重要组成, 可对土壤物质过程尤其是重金属环境行为产生重要影响[1]. Fe(Ⅱ)aq催化氧化铁晶相重组本质上是Fe(Ⅱ)aq与氧化铁中结构态Fe(Ⅲ)(Fe(Ⅲ)oxide)之间的电子转移与原子交换过程, 这一过程在20世纪80年代末已被发现[2], 但是直到近10年, 瑞典和美国的研究学者才通过稳定铁同位素示踪和光谱学等研究方法予以证实[3].近年来, 美国爱荷华大学Scherer教授课题组[1]采用稳定铁同位素57Fe-穆斯堡尔谱手段, 系统研究了Fe(Ⅱ)aq催化作用下不同氧化铁晶相重组过程, 其结果显示, 在Fe(Ⅱ)aq共存下, 氧化铁中的Fe(Ⅲ)oxide与Fe(Ⅱ)aq发生Fe原子交换, Fe(Ⅱ)aq进入氧化铁晶体表面或结构中被氧化成Fe(Ⅲ), 作为转变后二次氧化铁矿物的结构组成, 而氧化铁中某些Fe(Ⅲ)oxide则被还原释放到溶液中去, 这一过程也影响共存重金属的吸附/固定/释放等环境行为.美国太平洋西北国家实验室(PNNL) Rosso教授及其合作者[4]研究Fe(Ⅱ)aq催化下结晶度相对较高的α-Fe2O3的晶体变化时发现, 反应过程中Fe(Ⅱ)aq吸附到氧化铁表面后, 给出的电子可注入到氧化铁内层结构, 使氧化铁中Fe(Ⅲ)oxide发生还原释放, 而Fe(Ⅱ)aq给出电子后则在α-Fe2O3表面生长为新的α-Fe2O3.
Fe(Ⅱ)aq催化氧化铁晶相转变过程受反应条件影响较大. pH值是化学反应最重要的影响因素, Fe(Ⅲ)oxide与Fe(Ⅱ)aq之间的反应也受pH的影响:体系pH值越高, Fe(Ⅱ)更容易吸附到氧化铁表面, 而Fe(Ⅱ)在氧化铁表面的吸附率是影响Fe(Ⅱ)aq与Fe(Ⅲ)oxide之间Fe原子交换率的一个重要因素, 较高的Fe(Ⅱ)吸附率可促进反应[5], 因此, 体系pH值越高, Fe(Ⅱ)aq催化氧化铁晶相重组速率越快. Fe(Ⅱ)aq的初始浓度影响与氧化铁表面接触的Fe(Ⅱ)量, 进而影响其反应速率:在低Fe(Ⅱ)aq初始浓度范围内, 随着Fe(Ⅱ)在氧化铁表面吸附率的提高, 增加了与氧化铁表面Fe(Ⅲ)接触的吸附态Fe(Ⅱ)量, 可促进Fe(Ⅱ)-Fe(Ⅲ)之间的电子转移, 从而提高了它们之间的Fe原子交换率[4a, 5]; 但是, 在Fe(Ⅱ)aq初始浓度过高的条件下, 尤其是吸附的Fe(Ⅱ)量超过了氧化铁表面单层吸附位覆盖量, 会导致氧化铁表面电势失去梯度效应[4], 极大降低其中的电子转移速率.共存重金属离子也是影响Fe(Ⅱ)aq催化氧化铁晶相转变过程的重要因素:氧化铁结构中其他金属离子的Fe结构位取代, 可提高氧化铁Fe—O键的稳定性, 从而降低氧化铁晶相转变速率[6]; 共存游离态重金属影响下Fe(Ⅱ)aq催化氧化铁晶相转变过程的研究较少, 初步研究表明, 共存重金属可在一定程度上降低Fe(Ⅱ)aq催化氧化铁晶相转变速率, 同时, 部分重金属被固定到转变后的氧化铁结构中[6a, 7].
水铁矿是亚铁矿化过程中形成的初始氧化铁矿物, 是一种弱结晶的铁氢氧化物, 粒径较小, 具有极大的比表面积和高表面活性, 对土壤重金属等污染元素具有较大的吸附容量, 是影响土壤元素环境行为的重要土壤矿物[8].与其它高结晶度的氧化铁相比, 水铁矿的热力学稳定性较低, 在土壤中易通过重结晶, 转化为纤铁矿、针铁矿及磁铁矿等结晶度更高的氧化铁矿物[9].非表层土壤的厌氧条件下, 共存的由异化铁还原产生的Fe(Ⅱ)aq是水铁矿晶相转变/二次矿化的重要驱动力[10].相对于单纯的土壤铁循环, Fe(Ⅱ)aq催化作用下水铁矿的晶相转变具有更重要的环境化学意义, 水铁矿晶相转变过程中, Fe(Ⅱ)aq与不同反应时间段形成的氧化铁之间的进一步反应, 能对共存重金属污染物的吸附和共沉淀等环境行为产生重要影响[7, 11].通常情况下, 氧化铁溶解与重结晶被认为是结晶度较低的氧化铁转变形成结晶度较高氧化铁的反应过程机制[12].但是, 共存Fe(Ⅱ)aq作用下水铁矿二次矿化转变为更稳定氧化铁晶体的过程机制、尤其是在土壤重金属等环境条件影响下的转变过程, 仍有待深入研究.
因此, 本研究以水铁矿这一亚铁矿化过程中的初始形态氧化铁矿物为对象, 系统研究Fe(Ⅱ)aq作用下水铁矿的晶相转变过程以及土壤污染重金属Pb共存下反应过程变化及Pb的环境行为.采用稳定Fe同位素示踪研究Fe(Ⅱ)aq与水铁矿作用过程中的反应机制; 采用X射线衍射定量解析水铁矿晶相转变过程途径及Pb对转变产物的影响; 重点研究Fe(Ⅱ)aq作用下水铁矿晶相转变过程中Pb赋存形式的变换.以期通过以上所述研究, 系统揭示土壤Fe(Ⅱ)aq催化水铁矿晶相转变过程中Pb的环境行为机制.
2 结果与讨论
2.1 Fe(Ⅱ)催化水铁矿晶相转变及Pb的影响
在较为温和的反应条件下, 水铁矿结构相对稳定[9a], 本研究的前期实验表明, 在无Fe(Ⅱ)和Pb(Ⅱ)的对照体系中, 水铁矿的矿物相在研究的60 d内可保持稳定.而厌氧体系中, 1.0 mmol/L初始Fe(Ⅱ)aq作用下, 水铁矿能发生明显的晶相转变.如图 1(A)所示, 在反应进行到2 d时, 部分水铁矿转变为纤铁矿和针铁矿, 随着反应的进行, 纤铁矿比例降低, 而针铁矿比例则逐渐升高, 同时衍射图谱中出现磁铁矿衍射峰, 说明部分转变为磁铁矿.而在1.0 mmol/L的Pb(Ⅱ)影响下, Fe(Ⅱ)催化氧化铁晶相转变过程发生明显改变, 由图 1(B)可看出, 反应进行到2 d时, 部分水铁矿转变为纤铁矿和针铁矿, 形成微弱的纤铁矿和针铁矿衍射峰, 在反应进行到10 d后, 观察到微弱磁铁矿衍射峰出现.但是, 在Pb(Ⅱ)影响下, Fe(Ⅱ)aq催化氧化铁晶相转变在反应进行到15 d时水铁矿才全部转变为其他氧化铁矿物.从以上结果分析可知, Fe(Ⅱ)aq作用下, 水铁矿首先转变为纤铁矿, 生成的纤铁矿在Fe(Ⅱ)aq作用下, 进一步转变为针铁矿, 随着反应的进行, 部分纤铁矿或针铁矿转变为磁铁矿, 这也与Fe(Ⅱ)aq作用下氧化铁由低结晶度向高结晶度矿物相转变的趋势一致[1, 10b].而Pb(Ⅱ)的存在抑制Fe(Ⅱ)催化水铁矿晶相转变过程, Pb(Ⅱ)影响下, 水铁矿晶相转变速率明显降低, 同时, Fe(Ⅱ)aq作用的反应势降低, 纤铁矿进一步转化为其他矿物相的效率低于无Pb(Ⅱ)体系. Pb(Ⅱ)的存在也影响Fe(Ⅱ)aq催化水铁矿转变过程中转化产物的形貌.反应前水铁矿透射电镜表征(TEM)结果显示(此处TEM照片未提供), 水铁矿成多孔的絮状, 整体粒径约100 nm[7].不同条件下水铁矿转化后的形貌, 与其转化后形成的产物密切相关.在无Pb(Ⅱ)影响下, Fe(Ⅱ)aq催化水铁矿反应60 d时, 转化产物形貌呈规则性短棒状, 与针铁矿和磁铁矿的混合物相形貌一致[9a]; 而在Pb(Ⅱ)影响下, 反应60 d后样品形貌呈现典型的纤铁矿条状.
图 1
水铁矿晶相转变过程中的XRD图谱: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系(F:水铁矿; L:纤铁矿; G:针铁矿; M:磁铁矿)
Figure 1.
XRD patterns of the transformed ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)(F: ferrihydrite; L: lepidocrocite; G: Goethite; M: magnetite)
为进一步研究Pb(Ⅱ)对Fe(Ⅱ)催化水铁矿晶相转变过程的影响程度, 我们采用全谱分析软件Total Pattern Solution (TOPAS)对图 1中不同反应时间点的衍射数据进行矿物相组成的定量分析, 结果如图 2所示.从定量分析结果可以看出, 在反应进行到2 d时, 无Pb(Ⅱ)体系中78.4%的水铁矿转化为其他矿物相, 其中46.2%转化为纤铁矿, 22.6%转化为针铁矿, 9.6%转化为磁铁矿; 而在Pb(Ⅱ)影响下, 水铁矿晶相转化速率降低, 只有57.4%的水铁矿发生晶相转变, 大部分转化为纤铁矿(48.5%), 少量转化为针铁矿(8.9%), 无磁铁矿生成.反应进行到5 d时, 在无Pb(Ⅱ)影响体系中, 水铁矿全部转变成其他含铁矿物相, 纤铁矿、针铁矿和磁铁矿的比例分别为31.2%、55.9%和12.9%;而在Pb(Ⅱ)影响下, 只有67.2%的水铁矿转变为纤铁矿和针铁矿; 在后续的Fe(Ⅱ)催化作用过程中, 无Pb(Ⅱ)影响体系中纤铁矿逐步转变为针铁矿和磁铁矿, 在反应进行到10 d时纤铁矿全部转化为针铁矿和磁铁矿.而在Pb(Ⅱ)共存体系中, 直到10 d时, 转化产物才开始出现磁铁矿(6.5%), 反应进行到15 d时, 水铁矿才全部转化为其他氧化铁矿物相, 在研究的反应时间段60 d内, 水铁矿转化的初级产物纤铁矿是主要的二次矿物相.
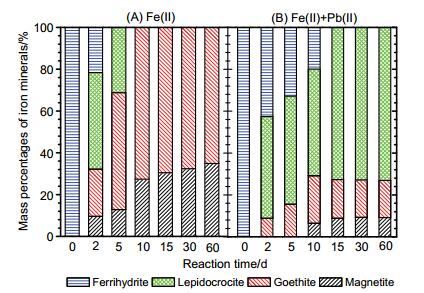 图 2
水铁矿晶相转变产物中氧化铁物相组成比例: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 2.
The compositions of iron oxides of the transformed ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)
图 2
水铁矿晶相转变产物中氧化铁物相组成比例: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 2.
The compositions of iron oxides of the transformed ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)
2.2 Fe(Ⅱ)催化水铁矿晶相转变过程中Pb的吸附固定
Fe(Ⅱ)aq催化氧化铁晶相转变过程可对重金属的环境行为产生重要影响:重金属结构取代的氧化铁, 在Fe(Ⅱ)aq催化晶相转变过程中, 部分重金属被释放[6a, 13]; 共存的游离态重金属, 则会在氧化铁晶相转变过程中被吸附, 并进一步进入转变后的氧化铁结构中被固定, 降低重金属活性[6a, 14].水铁矿作为亚铁矿化过程中形成的初始铁(氧)化物, 比表面积和结构活性较高, 对Pb(Ⅱ)具有较强的吸附性能.如图 3A所示, 在无Fe(Ⅱ)aq作用下, Pb(Ⅱ)在水铁矿表面发生较快的吸附, 在反应进行到10 d时, 吸附的Pb(Ⅱ)的浓度达到0.83 mmol/L, 其后趋于稳定, 但是无结构态Pb形成. Fe(Ⅱ)aq催化水铁矿晶相转变过程则对Pb的赋存形式产生重要影响. 图 3B为Fe(Ⅱ)催化水铁矿晶相转变过程中不同形式Pb浓度的变化.在Fe(Ⅱ)aq催化水铁矿晶相转变过程中, 溶液中游离态Pb(Ⅱ)浓度逐渐下降, 在反应进行到15 d时下降到0.23 mmol/L; 相应的, 吸附态Pb(Ⅱ)浓度逐渐上升, 在反应进行到5 d时达到0.44 mmol/L, 进一步反应其浓度减低, 在15 d时稳定在(0.15±0.02) mmol/L; 结构态Pb浓度在反应过程中则稳定升高, 在15 d时达到0.63 mmol/L, 其后浓度值趋于稳定.
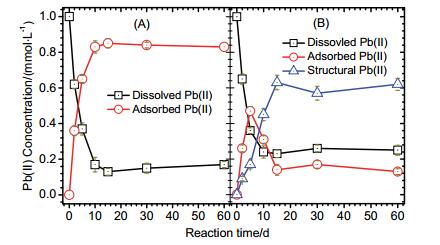 图 3
无Fe(Ⅱ)作用时水铁矿对Pb(Ⅱ)的吸附(A), 及Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中Pb不同赋存形态的浓度变化(B)
Figure 3.
The adsorption of Pb(Ⅱ) on the surface of ferrihydrite (A) and the concentrations of different Pb(Ⅱ) species during the Fe(Ⅱ)-catalyzed phase transformation of ferrihydrite with the coexistence of Pb(Ⅱ) (B)
图 3
无Fe(Ⅱ)作用时水铁矿对Pb(Ⅱ)的吸附(A), 及Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中Pb不同赋存形态的浓度变化(B)
Figure 3.
The adsorption of Pb(Ⅱ) on the surface of ferrihydrite (A) and the concentrations of different Pb(Ⅱ) species during the Fe(Ⅱ)-catalyzed phase transformation of ferrihydrite with the coexistence of Pb(Ⅱ) (B)
从图 3结果可知, 水铁矿对Pb(Ⅱ)具有较强的吸附性能. Fe(Ⅱ)aq催化水铁矿晶相转变形成的含不同种类氧化铁的混合物相, 对Pb(Ⅱ)也具有较强的吸附性能, 在这一过程中, 溶液态Pb(Ⅱ)逐步转变为吸附态Pb(Ⅱ), 而吸附到氧化铁表面的Pb(Ⅱ), 可进一步在水铁矿晶相转变过程中, 被固定到转变后形成的氧化铁结构中.结构化固定可能的机理为Pb取代Fe结构位, 或Pb在转变后的氧化铁中, 被氧化铁晶体包裹[1].
为进一步对Fe(Ⅱ)aq催化水铁矿晶相转变过程中Pb(Ⅱ)的固定进行验证, 我们对反应到30 d时无Pb(Ⅱ)和有Pb(Ⅱ)时Fe(Ⅱ)aq催化水铁矿晶相转变体系固态样品采用Fe的K-edge进行同步辐射X射线吸收精细结构光谱(EXAFS)表征(图 4).在Pb(Ⅱ)共存的Fe(Ⅱ)aq催化水铁矿晶相转变体系中, Fe—O键的吸收峰相对于无Pb(Ⅱ)体系样品发生负移. 1.53 处的吸收峰为最靠近氧原子处的八配位Fe或四配位Fe散射产生, 可作为样品第一层壳Fe—O键距指标.在Pb(Ⅱ)共存体系中, 晶相转变后形成的氧化铁二次矿物中, Fe—O间距相比于无Pb(Ⅱ)体系形成的氧化铁二次矿物有一定降低.而有报道指出, 如果氧化铁结构中存在其它金属离子元素(如Cd、Pb、Zn、Mn、Al), 氧化铁第一层壳的Fe—O间距将相应减小[15].因此, 图 4研究结果结合所述的文献报道, 可进一步说明, Pb(Ⅱ)共存情况下Fe(Ⅱ)aq催化水铁矿晶相转变过程中Pb被固定到转变后的二次氧化铁矿物结构中.但是, 其结构化固定的Fe结构位取代或晶格包裹机制、及两种固定机制对Pb固定的比例贡献, 从本研究现有的数据还无法确认, 还需进一步研究.
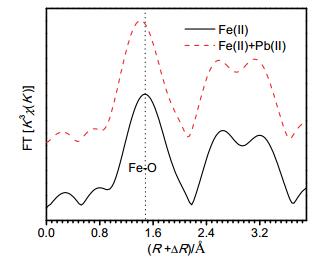 图 4
无Pb(Ⅱ)和Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中转化产物的EXAFS图谱
Figure 4.
The Fourier transform FT of K3 weighted EXAFS spectra (Fe K-edge) as a radial structure function without the phase shift correction for the Fe(Ⅱ)-catalyzing transformed ferrihydrite with or without coexistence of Pb(Ⅱ)
图 4
无Pb(Ⅱ)和Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中转化产物的EXAFS图谱
Figure 4.
The Fourier transform FT of K3 weighted EXAFS spectra (Fe K-edge) as a radial structure function without the phase shift correction for the Fe(Ⅱ)-catalyzing transformed ferrihydrite with or without coexistence of Pb(Ⅱ)
2.3 Pb(Ⅱ)影响下Fe(Ⅱ)催化水铁矿晶相转变过程中Fe(Ⅱ)物种分布
为进一步探明Pb(Ⅱ)影响下Fe(Ⅱ)催化水铁矿晶相转变过程机制, 本研究对反应过程中直接的作用物种Fe(Ⅱ)在溶液、氧化铁矿物表面及氧化铁结构中(包括转化后形成的磁铁矿的分子组成Fe(Ⅱ))的浓度分布进行了研究, 结果如图 5所示.在无Pb(Ⅱ)和有Pb(Ⅱ)的体系中, 反应过程中溶解态Fe(Ⅱ)浓度均呈现较快的下降趋势, 无Pb(Ⅱ)影响体系在反应进行到第15 d后达到稳定, Pb(Ⅱ)影响体系在第10 d达到稳定; 而吸附态Fe(Ⅱ)浓度在反应初始阶段快速增加, 然后在反应2 d后逐步降低; 结构态Fe(Ⅱ)浓度在反应初始阶段快速增加, 在反应15 d后趋于稳定. Pb(Ⅱ)的共存, 对Fe(Ⅱ)物种的分布存在重要的影响, 在Pb共存体系中, 由于Pb(Ⅱ)的竞争性吸附, 溶解态Fe(Ⅱ)浓度相对于无Pb(Ⅱ)体系浓度下降慢, 在反应进行到10 d时, Pb(Ⅱ)共存体系中Fe(Ⅱ)下降到0.51 mmol/L, 无Pb(Ⅱ)体系中溶解态Fe(Ⅱ)则下降到0.32 mmol/L.同样由于Pb(Ⅱ)与Fe(Ⅱ)对水铁矿及其转变后的氧化铁表面位和结构位的竞争性关系, Pb(Ⅱ)共存体系中吸附态Fe(Ⅱ)和结构态Fe(Ⅱ)的浓度均低于无Pb(Ⅱ)反应体系.需要指出的是, 从图 1结果我们已知, 在无Pb(Ⅱ)影响的Fe(Ⅱ)aq催化水铁矿晶相转变体系中, 转变后的矿物相中含有磁铁矿组分, 因此, 图 5中无Pb(Ⅱ)体系中的结构态Fe(Ⅱ)包含了磁铁矿分子结构中的Fe(Ⅱ).
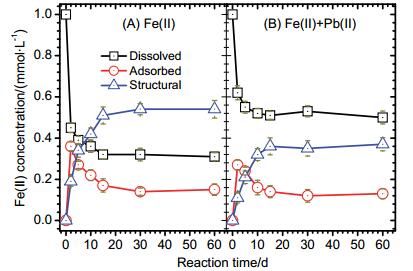 图 5
Fe(Ⅱ)催化水铁矿晶相转变过程中不同分布的总Fe(Ⅱ)物种: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 5.
The concentrations of different total Fe(Ⅱ) species during the Fe(Ⅱ)-catalyzed transformation of ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)
图 5
Fe(Ⅱ)催化水铁矿晶相转变过程中不同分布的总Fe(Ⅱ)物种: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 5.
The concentrations of different total Fe(Ⅱ) species during the Fe(Ⅱ)-catalyzed transformation of ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)
Fe(Ⅱ)物种的存在形式, 直接影响水铁矿晶相转变过程.在无Pb(Ⅱ)存在情况下, 吸附到水铁矿及其转变后的氧化铁表面的Fe(Ⅱ)增加, 部分吸附的Fe(Ⅱ)通过氧化铁矿物晶体的闭合或原子交换进入到氧化铁结构中[1, 16], 同时, 水铁矿晶相发生转变, 形成二次矿物, 如纤铁矿、针铁矿、磁铁矿(图 1).而在Pb(Ⅱ)共存体系中, 由于Pb(Ⅱ)的竞争, 氧化铁表面和结构中Fe(Ⅱ)的量均低于无Pb(Ⅱ)体系中氧化铁表面和结构中的量. Boland等[17]指出, 与氧化铁直接关联的Fe(Ⅱ)物种(包括吸附态和结构态)是影响Fe(Ⅱ)aq催化氧化铁晶相转变的重要因素.在有Pb(Ⅱ)共存情况下, 降低了氧化铁直接相关的Fe(Ⅱ)物种量, 因此, 使水铁矿晶相转变过程受到抑制, 水铁矿的转变速率降低, 转变的途径也发生改变.
2.4 Pb(Ⅱ)影响下Fe(Ⅱ)催化水铁矿晶相转变的Fe原子交换抑制机制
通过以上的研究结果可知, Pb(Ⅱ)影响下, Fe(Ⅱ)催化水铁矿晶相转变速率受到抑制, 转化产物发生改变.游离态Fe(Ⅱ)与氧化铁中结构态Fe(Ⅲ)之间的Fe原子交换, 是Fe(Ⅱ)催化氧化铁晶相重组的主要反应机制[5, 18].游离态Fe(Ⅱ)吸附到氧化铁表面后, 给出电子, 自身被氧化成Fe(Ⅲ), 作为形成的二次氧化铁矿物结构组成, 给出的电子通过固态电子传导, 传递到氧化铁中其它位置的结构态Fe(Ⅲ), 使其发生还原, 进一步溶解释放到氧化铁表面或溶液中, 形成游离态Fe(Ⅱ).反应过程中, 体系游离态Fe(包括溶解态和吸附态)总量与结构态Fe量均没有发生变化, 但是游离态Fe与结构态Fe发生了交换, 交换过程中氧化铁晶相发生转变[19].同时, 正是由于在反应过程中游离态Fe(Ⅱ)与结构Fe(Ⅲ)的量均没有发生变化, 因此, 该过程可被认为是催化反应.
为进一步阐明Pb(Ⅱ)对Fe(Ⅱ)aq催化水铁矿晶相转变过程的影响机制, 本文采用稳定Fe同位素示踪研究反应过程中Fe原子交换过程.以57Fe标记的Fe(Ⅱ)溶液与Fe稳定同位素自然丰度组成的水铁矿进行反应, 通过反应过程中57Fe在游离态和结构态之间的组成比例变化, 研究Fe原子交换速率.通过获得的反应过程中体系四种Fe稳定同位素组成比例的数据, 采用公式(1) 计算Fe原子交换率[5, 20], 其中Naq代表反应体系中总的游离态Fe(Ⅱ)数量, NFhTot代表水铁矿固态中总Fe数量, ƒaqi代表反应开始前体系中游离态Fe(Ⅱ)同位素组成, ƒFhi代表反应开始前体系中水铁矿中Fe同位素组成, ƒFe(Ⅱ)t代表在反应时间t时游离态Fe(Ⅱ)的稳定同位素组成:
图 6为Fe(Ⅱ)aq催化水铁矿晶相转变体系中游离态54Fe、56Fe和57Fe的组成变化过程, 其中图 6(A)为无Pb(Ⅱ)共存的反应体系, 图 6(B)为Pb(Ⅱ)共存时的反应体系(Fe的四种稳定同位素中, 58Fe的自然比例仅为0.28%, 为避免实验及分析过程中可能的误差影响, 在这里f58Fe变化的数据没有单独给出进行讨论).在反应开始前, 两个体系中Fe(Ⅱ)均为标记的57Fe试剂的比例组成, f57Fe为0.964, f54Fe和f56Fe分别为0.00016和0.013.随着反应的进行, 两个体系中57Fe组成比例均逐渐下降, 54Fe和56Fe的组成比例则均逐渐上升.在反应进行到60 d时, 无Pb(Ⅱ)影响的体系中f57Fe下降到0.109, Pb(Ⅱ)共存反应体系中f57Fe下降到0.359.相应的, 无Pb(Ⅱ)体系f54Fe和f56Fe分别上升到0.037和0.851, Pb(Ⅱ)共存体系f54Fe和f56Fe分别上升到0.028和0.611.这一结果说明, 游离态Fe(Ⅱ)与水铁矿共存体系中, 有Pb(Ⅱ)和无Pb(Ⅱ)影响体系中, 均有部分游离态的57Fe进入到氧化铁结构, 而氧化铁中同位素自然丰度组成的Fe(Ⅲ), 则部分被还原释放到溶液中, 游离态Fe(Ⅱ)与水铁矿中的结构态Fe(Ⅲ)在两个体系中均能发生Fe原子交换.但是在Pb(Ⅱ)存在的体系中, f57Fe下降的程度, 低于无Pb(Ⅱ)影响体系的f57Fe, 相应的, f54Fe和f56Fe上升的比例, 高于无Pb(Ⅱ)影响体系的f57Fe.这一结果说明, 在Pb(Ⅱ)共存体系中, 较少的游离态57Fe进入到氧化铁, 从氧化铁中释放到溶液中的结构态Fe(Ⅲ)量也减少, Fe原子交换发生相对较慢.为定量研究Pb(Ⅱ)对Fe(Ⅱ)aq催化水铁矿晶相转变过程中Fe原子交换速率的影响, 我们采用公式(1) 计算了反应过程中Fe原子的交换速率, 两个反应体系中Fe原子交换率变化动力学如图 7所示.从图 7结果可以看出, 两个体系中Fe原子交换速率均逐步提高, 同时, Pb(Ⅱ)共存影响下, Fe(Ⅱ)aq催化水铁矿晶相转变过程中Fe原子交换速率明显低于无Pb(Ⅱ)影响体系, 在反应进行到60 d时, 无Pb(Ⅱ)影响体系中Fe原子交换速率达到54.5%, 而有Pb(Ⅱ)共存时, Fe原子交换速率下降到37.3%.这一结果说明, Pb(Ⅱ)抑制了Fe(Ⅱ)aq催化水铁矿晶相转变过程中Fe原子的交换.
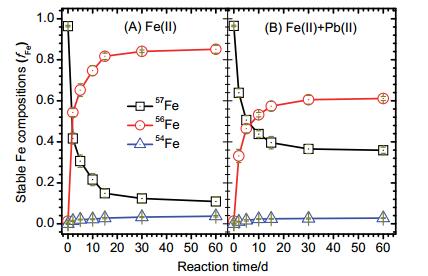 图 6
Fe(Ⅱ)催化水铁矿晶相转变过程中溶解态Fe(Ⅱ)的Fe稳定同位素组成比例变化: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 6.
The compositions of different stable Fe isotopes during the 57Fe(Ⅱ)-catalyzed transformation of ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)
图 6
Fe(Ⅱ)催化水铁矿晶相转变过程中溶解态Fe(Ⅱ)的Fe稳定同位素组成比例变化: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 6.
The compositions of different stable Fe isotopes during the 57Fe(Ⅱ)-catalyzed transformation of ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)
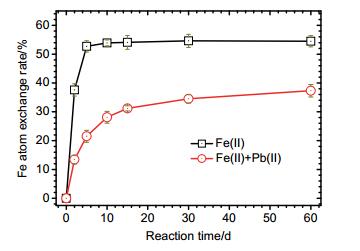 图 7
无Pb(Ⅱ)和Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中游离态Fe(Ⅱ)与结构态Fe(Ⅲ)之间Fe原子交换率
Figure 7.
The Fe atom exchange rates between Fe(Ⅱ)aq and structural Fe(Ⅲ) of ferrihydrite during the Fe(Ⅱ)-catalyzing transformation of ferrihydrite with or without coexistence of Pb(Ⅱ)
图 7
无Pb(Ⅱ)和Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中游离态Fe(Ⅱ)与结构态Fe(Ⅲ)之间Fe原子交换率
Figure 7.
The Fe atom exchange rates between Fe(Ⅱ)aq and structural Fe(Ⅲ) of ferrihydrite during the Fe(Ⅱ)-catalyzing transformation of ferrihydrite with or without coexistence of Pb(Ⅱ)
以以上研究结果为基础, 结合游离态Fe(Ⅱ)催化氧化铁晶相转变这一研究的相关文献报道, 我们对Fe(Ⅱ)催化水铁矿晶相转变过程中Pb的影响及其环境行为机制进行探讨, 其机理示意如图 8所示.在无Pb(Ⅱ)影响下, 体系中溶液态Fe(Ⅱ)在水铁矿表面发生充分的吸附, 吸附到水铁矿表面的Fe(Ⅱ)与水铁矿中的结构态Fe(Ⅲ)发生Fe原子交换, Fe(Ⅱ)给出一个电子, 自身被氧化成二次矿物的结构态Fe(Ⅲ), 而水铁矿中的某些位置的结构态Fe(Ⅲ)获得电子后, 被还原成Fe(Ⅱ), 释放到氧化铁表面或溶液中.同时, Fe原子交换中的电子转移过程, 也催化水铁矿发生晶相转变[19a].在无Pb(Ⅱ)影响体系中, 由于与氧化铁固体直接关联的Fe(Ⅱ)(包括溶液态和吸附态)的丰度较高, 因此, 水铁矿能发生较高速率的晶相转变, 形成了结晶度较高的针铁矿和磁铁矿等转变产物.而Pb(Ⅱ)影响的Fe(Ⅱ)aq催化水铁矿晶相转变体系中, 由于Pb(Ⅱ)与Fe(Ⅱ)对水铁矿表面吸附位的竞争性, 减低了吸附到水铁矿表面的Fe(Ⅱ)的量, 同时部分Pb(Ⅱ)吸附到水铁矿表面, 虽然较少的Fe(Ⅱ)量也能催化水铁矿晶相转变, 但是, 由于Pb(Ⅱ)占据水铁矿部分吸附位, 使Fe(Ⅱ)催化水铁矿晶相转变过程速率降低.同时, 在水铁矿晶相转变过程中, 部分Pb(Ⅱ)被固定到转变后形成的矿物相中, 其机制包括晶体包裹和结构位取代.重金属通常取代氧化铁结构中的八面体配位Fe(Ⅲ), 由于取代的Pb(Ⅱ)与原八面体配位Fe(Ⅲ)存在电荷差异, 导致形成的Pb(Ⅱ)结构取代氧化铁结构电荷失衡, 整体偏负[9a], 使其从Fe(Ⅱ)aq获得电子的效率降低, 从而降低氧化铁结构中的电子转移效率, 进一步抑制Fe(Ⅱ)催化氧化铁的晶相转变过程[6b].因此, 在Fe(Ⅱ)催化水铁矿晶相转变过程中, 共存的重金属Pb(Ⅱ)能降低水铁矿晶相转变速率, 同时, 部分Pb被固定到转变后的二次矿物结构中.
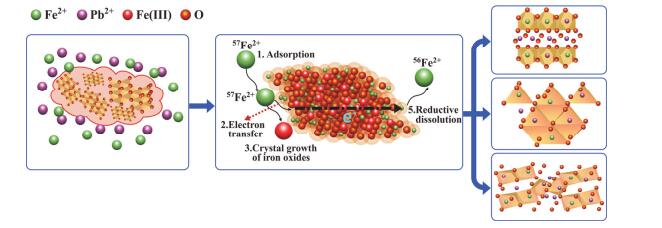 图 8
Pb(Ⅱ)影响下Fe(Ⅱ)催化水铁矿晶相转变过程机理示意图
Figure 8.
The mechanism diagram of Fe(Ⅱ)-catalyzed phase transformation of ferrihydrite with the coexistence of Pb(Ⅱ)
图 8
Pb(Ⅱ)影响下Fe(Ⅱ)催化水铁矿晶相转变过程机理示意图
Figure 8.
The mechanism diagram of Fe(Ⅱ)-catalyzed phase transformation of ferrihydrite with the coexistence of Pb(Ⅱ)
3 结论
厌氧体系中, 共存的游离态Fe(Ⅱ)可与水铁矿中结构态Fe(Ⅲ)发生Fe原子交换, 同时催化水铁矿发生晶相转变.共存的重金属离子Pb(Ⅱ)抑制游离态Fe(Ⅱ)与水铁矿中结构态Fe(Ⅲ)之间的Fe原子交换过程, 降低水铁矿晶相转变速率. Pb(Ⅱ)也影响Fe(Ⅱ)催化水铁矿晶相转变的产物, 在无Pb(Ⅱ)存在情况下, 水铁矿首先转化为纤铁矿, 然后进一步转化为针铁矿和磁铁矿; 而Pb(Ⅱ)影响下, 水铁矿晶相转变速率降低, 纤铁矿为主要产物, 少量转化为针铁矿和磁铁矿.在游离态Fe(Ⅱ)催化水铁矿晶相转变过程中, 共存的重金属离子Pb(Ⅱ)可通过晶格包裹或结构位取代机制被转变后的二次氧化铁矿物相结构化固定, 从而降低Pb(Ⅱ)离子活性.
4 实验部分
4.1 水铁矿制备
水铁矿的合成主要参考已有文献报道的方法[9c, 17].首先, 将20 g Fe(NO3)3•9H2O加入到250 mL的双蒸馏水中, 待完全溶解后采用浓度为1 mol/L的KOH调节溶液pH至7~8.产生的悬浮液重复性离心-双蒸馏水洗涤处理, 直至泥状物质pH达到约7.5, 即获得水铁矿悬浮液.水铁矿悬浮液在冰箱中冰冻保存, 在三天内用于实验.部分水铁矿悬浮液经离心与冷冻干燥后用于结构和形貌表征.
4.2 Fe(Ⅱ)aq催化水铁矿晶相转变实验
除特别说明外, 本实验均在厌氧手套箱中进行(Bactron Ⅱ, Shellab, 美国), 所有的反应溶液均以高纯N2除氧2 h以上后转移至厌氧箱, 并在厌氧箱中平衡48 h后使用. 100 mmol/L的Fe(Ⅱ)溶液储备液在厌氧箱中把FeCl2•9H2O溶于除氧后的双蒸馏水中制得. 100 mmol/L的Pb(Ⅱ)溶液在厌氧箱中把Pb(NO3)2溶于除氧后的双蒸馏水中制得. 57Fe标记的Fe(Ⅱ)溶液(简称57Fe(Ⅱ)溶液)由57Fe(0) 粉末(购自isoflex, 纯度>95.0%)溶解于4 mol/L的HCl, 然后进一步采用超纯水稀释获得100 mmol/L的57Fe(Ⅱ)储备液.
反应在15 mL的离心管中进行, 每个时间点设置3个平行的离心管反应器.每个离心管初始加入30 mmol/L的水铁矿(约3.2 g/L)、1.0 mmol/L的Fe(Ⅱ)溶液、以及0或1.0 mmol/L的Pb(Ⅱ).反应溶液由20 mmol/L的1, 4-哌嗪二乙磺酸(PIPES)的缓冲液控制反应pH值为6.5.待所有反应试剂加入到离心管后, 盖紧然后采用锡箔纸包裹避光, 放置于厌氧手套箱中的垂直振荡器上, 室温条件下振荡反应.反应进行到设定的时间点, 在厌氧手套箱中取样, 每次取样三个离心管平衡处理分析.首先, 将离心管移出厌氧手套箱, 在离心机中8000 r/min转速下离心15 min, 然后迅速再放入厌氧手套箱中进行下一步处理.在厌氧手套箱中进行离心后物质的固液分离, 液体采用针头过滤器(0.22 μm)过滤收集, 用于分析溶液态物质; 固体首先采用4 mL初始浓度为0.4 mol/L的HCl提取, 过滤后液体用于分析吸附态物质[21]; 剩下的固体加入4 mL初始浓度为4 mol/L的HCl进行溶解, 固态物质完全溶解后进一步过滤收集液体, 用于结构态物质含量分析.以上所述三种液体样品均进一步用于后续的Fe2+和总Fe浓度及Fe稳定同位素组成分析.为对反应后固态物质进行物化性质表征, 设置相同的反应体系, 反应后固液分离处理, 固体样品进行冷冻干燥, 用于后续的表征研究.
4.3 测试与表征
实验中的分析方法主要参考本研究组已发表的论文[18b].溶液、0.4 mol/L HCl提取液及固态物质溶解后液体中Fe2+和总Fe浓度采用传统的邻菲啰啉比色法进行分析[22], 分析仪器为紫外可见分光光度计(TU-1800, 北京普析通用).不同溶液中铁的四种稳定同位素组成(54Fe、56Fe、57Fe及58Fe)采用电感耦合等离子体质谱仪(PerkinElmer ICP-MS NX300) 的KED模式进行分析:待测溶液经自动进样器(采用玻璃罩与外界隔离)吸取、同心雾化器雾化、电离后经过三级采样锥进入碰撞池内反应.为避免同质元素16O40Ar对56Fe的干扰, 碰撞池中气体采用7%的H2+93%的He(纯度为99.999%), 气流量为20 PSI.反应过程中固态物质冷冻干燥后, 进行结构和性质表征.晶相组成的定量和定性分析采用X射线衍射(XRD)进行表征, 实验在台湾同步辐射中心(NSRRC) TLS光源的BL17A1光速线基站进行: TLS环能量1.5 GeV, 常用电流360 mA; X射线测试波长1.321863 (≈9.3 keV), 由超导波长转换磁体和Si(111) 三角形晶体传导; 衍射数据采用Mar345成像板探测器收集, 成像板探测器距离样品位180 mm, 与样品正面暴露持续时间5 min; 衍射角根据LaB6标准的布拉格位置进行校准.衍射数据的定性分析根据国际衍射数据库中心的粉末衍射标准数据库(ICDD PDF-2, Release 2008) 进行[23].水铁矿理论结构包含20%的四面体配位铁和80%的八面体配位铁[24], 但是由于其结晶度低, 在表征中作为无定形组分.为了同时定量分析固体中结晶态和无定形组分, 所有的样品在表征前均加入20%(质量比)的CaF2 (449717-25G, Merck, 德国)作为内标, 衍射数据采用TOPAS程序进行Rietveld定量分析[25].水铁矿及其反应后固体样品的形貌及粒度采用透射电子电镜(TEM)表征, 仪器为Philips-CM12透射电子电镜, 操作条件为200 kV[7].
-
-
[1]
Latta, D. E.; Bachman, J. E.; Scherer, M. M. Environ. Sci. Technol. 2012, 46, 10614. doi: 10.1021/es302094a
-
[2]
(a) Suter, D.; Siffert, C.; Sulzberger, B.; Stumm, W. Nturwissenschaften 1988, 75, 571. (b) Suter, D.; Banwart, S.; Stumm, W. Langmuir 1991, 7, 809.
-
[3]
(a) Williams, A. G.; Scherer, M. M. Environ. Sci. Technol. 2004, 38, 4782. (b) Pedersen, H. D.; Postma, D.; Jakobsen, R.; Larsen, O. Geochim. Cosmochim. Acta 2005, 69, 3967.
-
[4]
(a) Yanina, S. V.; Rosso, K. M. Science 2008, 320, 218. (b) Rosso, K. M.; Yanina, S. V.; Gorski, C. A.; Larese-Casanova, P.; Scherer, M. M. Environ. Sci. Technol. 2010, 44, 61.
-
[5]
Frierdich, A. J.; Helgeson, M.; Liu, C.; Wang, C.; Rosso, K. M.; Scherer, M. M. Environ. Sci. Technol. 2015, 49, 8479. doi: 10.1021/acs.est.5b01276
-
[6]
(a) Frierdich, A. J.; Catalano, J. G. Environ. Sci. Technol. 2012, 46, 11070. (b) Frierdich, A. J.; Scherer, M. M.; Bachman, J. E.; Engelhard, M. H.; Rapponotti, B. W.; Catalano, J. G. Environ. Sci. Technol. 2012, 46, 10031.
-
[7]
Liu, C. S.; Zhu, Z. K.; Li, F. B.; Liu, T. X.; Liao, C. Z.; Lee, J. J.; Shih, K. M.; Tao, L.; Wu, Y. D. Chem. Geol. 2016, 444, 110. doi: 10.1016/j.chemgeo.2016.10.002
-
[8]
(a) Jambor, J. L. ; Dutrizac, J. E. Chem. Rev. 1998, 98, 2549. (b) Wang, X. M. ; Li, W. ; Harrington, R. ; Liu, F. ; Parise, J. B. ; Feng, X. H. ; Sparks, D. L. Environ. Sci. Technol. 2013, 47, 10322. (c) Liu, J. ; Yang, J. ; Zeng, X. ; Wang, J. ; Sparks, D. Acta Chim. Sinica 2017, DOI: 10.6023/A17030082.(刘瑾, 杨建军, 曾希柏, JianWang, DonaldSparks, 化学学报, 2017, DOI:10.6023/A17030082.); (d)Wang, X.;Peng, J.;Xu, H.;Tan, W.;Liu, F.;Huang, Q.;Feng, X.ActaChim.Sinica2017, DOI:10.6023/A17020046.(王小明, 彭晶, 徐欢欢, 谭文峰, 刘凡, 黄巧云, 冯雄汉, 化学学报, 2017, DOI:10.6023/A17020046.)
-
[9]
(a) Cornell, R. M. ; Schwertmann, U. The Iron Oxides: Structure, Properties, Reactions, Occurences and Uses, 2nd ed. , Wiley-VCH, Weinheim, 2003. (b) Wang, X. ; Liu, H. ; Wei, Y. ; Li, Y. ; Zhao, X. ; Wang, Y. Acta Chim. Sinica 2008, 66, 685. (王欣, 刘辉, 魏雨, 李彦涛, 赵新强, 王延吉, 化学学报, 2008, 66, 685. ); (c) Das, S. ; Hendry, M. J. ; Essilfie-Dughan, J. Environ. Sci. Technol. 2011, 45, 5557. (d) Wang, X. M. ; Zhu, M. Q. ; Lan, S. ; Ginder-Vogel, M. ; Liu, F. ; Feng, X. H. Chem. Geol. 2015, 415, 37.
-
[10]
(a) Tronc, E.; Belleville, P.; Jolivet, J. P.; Livage, J. Langmuir 1992, 8(1), 313. (b) Hansel, C. M.; Benner, S. G.; Fendorf, S. Environ. Sci. Technol. 2005, 39(18), 7147.
-
[11]
(a) Stewart, B. D.; Nico, P. S.; Fendorf, S. Environ. Sci. Technol. 2009, 43(13), 4922. (b) Amstaetter, K.; Borch, T.; Larese-Casanova, P.; Kappler, A. Environ. Sci. Technol. 2010, 44(1), 102. (c) Felmy, A. R.; Moore, D. A.; Rosso, K. M.; Qafoku, O.; Rai, D.; Buck, E. C.; Ilton, E. S. Environ. Sci. Technol. 2011, 45(9), 3952.
-
[12]
(a) Schwertmann, U.; Taylor, R. M. Clays Clay Miner. 1972, 20(3), 151. (b) Yang, L.; Steefel, C. I.; Marcus, M. A.; Bargar, J. R. Environ. Sci. Technol. 2010, 44(14), 5469.
-
[13]
Frierdich, A. J.; Catalano, J. G. Environ. Sci. Technol. 2012, 46, 1519. doi: 10.1021/es203272z
-
[14]
(a) Wu, Z. ; Zhang, W. ; Sun, Z. Acta Chim. Sinica 2010, 68, 769. (吴震生, 张卫民, 孙中溪, 化学学报, 2010, 68, 769. ); (b) Cao, F. ; Wu, Y. ; Liu, H. ; Wei, Y. Acta Chim. Sinica 2008, 66, 1405. (曹付玲, 吴育飞, 刘辉, 魏雨, 化学学报, 2008, 66, 1405. )
-
[15]
(a) Alvarez, M.; Rueda, E. H.; Sileo, E. E. Geochim. Cosmochim. Acta 2007, 71, 1009. (b) Kaur, N.; Gräfe, M.; Singh, B.; Kennedy, B. Clays Clay Miner. 2009, 57(2), 234.
-
[16]
Jang, J. H.; Dempsey, B. A.; Catchen, G. L.; Burgos, W. D. Colloids Surf., A 2003, 221(1), 55. http://www.sciencedirect.com/science/article/pii/S0927775703001341
-
[17]
Boland, D. D.; Collins, R. N.; Miller, C. J.; Glover, C. J.; Waite, T. D. Environ. Sci. Technol. 2014, 48(16), 9086. doi: 10.1021/es501750z
-
[18]
(a) Gorski, C. A. ; Handler, R. M. ; Beard, B. ; Pasakarnis, T. ; Johnson, C. M. ; Scherer, M. M. Environ. Sci. Technol. 2012, 46, 12399. (b) Liu, C. S. ; Wei, Z. Q. ; Li, F. B. ; Dong, J. Scientia Sinica Terrae 2016, 46, 1542. (刘承帅, 韦志琦, 李芳柏, 董军, 中国科学: 地球科学, 2016, 46, 1542. )
-
[19]
(a) Handler, R. M.; Beard, B. L.; Johnson, C. M.; Scherer, M. M. Environ. Sci. Technol. 2009, 43, 1102. (b) Joshi, P.; Gorski, C. A. Environ. Sci. Technol. 2016, 50, 7315.
-
[20]
Handler, R. M.; Frierdich, A. J.; Johnson, C. M.; Rosso, K. M.; Beard, B. L.; Wang, C.; Latta, D. E.; Neumann, A.; Pasakarnis, T.; Premaratne, W. A. P. J.; Scherer, M. M. Environ. Sci. Technol. 2014, 48, 11302. doi: 10.1021/es503084u
-
[21]
Reddy, T. R.; Frierdich, A. J.; Beard, B. L.; Johnson, C. M. Chem. Geol. 2015, 397, 118. doi: 10.1016/j.chemgeo.2015.01.018
-
[22]
Schilt, A. A. Applications of 1, 10-Phenanthroline and Related Compounds, 1st ed., Pergamon Press, Oxford, 1969.
-
[23]
Lu, X. W.; Shih, K. M.; Liu, C. S.; Wang, F. Environ. Sci. Technol. 2013, 47, 9972. doi: 10.1021/es401674d
-
[24]
Michel, F. M.; Ehm, L.; Antao, S. M.; Lee, P. L.; Chupas, P. J.; Liu, G.; Strongin, D. R.; Schoonen, M. A. A.; Phillips, B. L.; Parise, J. B.; Science 2007, 316, 1726. doi: 10.1126/science.1142525
-
[25]
(a) De La Torre, A. G.; Bruque, S.; Aranda, M. A. G. J. Appl. Crystallogr. 2001, 34, 196. (b) Bernasconi, A.; Dapiaggi, M.; Gualtieri, A. F. J. Appl. Crystallogr. 2014, 47, 136.
-
[1]
-

图 1 水铁矿晶相转变过程中的XRD图谱: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系(F:水铁矿; L:纤铁矿; G:针铁矿; M:磁铁矿)
Figure 1 XRD patterns of the transformed ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)(F: ferrihydrite; L: lepidocrocite; G: Goethite; M: magnetite)

图 2 水铁矿晶相转变产物中氧化铁物相组成比例: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 2 The compositions of iron oxides of the transformed ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)

图 3 无Fe(Ⅱ)作用时水铁矿对Pb(Ⅱ)的吸附(A), 及Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中Pb不同赋存形态的浓度变化(B)
Figure 3 The adsorption of Pb(Ⅱ) on the surface of ferrihydrite (A) and the concentrations of different Pb(Ⅱ) species during the Fe(Ⅱ)-catalyzed phase transformation of ferrihydrite with the coexistence of Pb(Ⅱ) (B)

图 4 无Pb(Ⅱ)和Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中转化产物的EXAFS图谱
Figure 4 The Fourier transform FT of K3 weighted EXAFS spectra (Fe K-edge) as a radial structure function without the phase shift correction for the Fe(Ⅱ)-catalyzing transformed ferrihydrite with or without coexistence of Pb(Ⅱ)

图 5 Fe(Ⅱ)催化水铁矿晶相转变过程中不同分布的总Fe(Ⅱ)物种: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 5 The concentrations of different total Fe(Ⅱ) species during the Fe(Ⅱ)-catalyzed transformation of ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)

图 6 Fe(Ⅱ)催化水铁矿晶相转变过程中溶解态Fe(Ⅱ)的Fe稳定同位素组成比例变化: (A) Fe(Ⅱ)催化体系; (B) Pb(Ⅱ)影响下的Fe(Ⅱ)催化体系
Figure 6 The compositions of different stable Fe isotopes during the 57Fe(Ⅱ)-catalyzed transformation of ferrihydrite: (A) Catalyzed by Fe(Ⅱ); (B) Catalyzed by Fe(Ⅱ) with the coexistence of Pb(Ⅱ)

图 7 无Pb(Ⅱ)和Pb(Ⅱ)共存体系Fe(Ⅱ)催化水铁矿晶相转变过程中游离态Fe(Ⅱ)与结构态Fe(Ⅲ)之间Fe原子交换率
Figure 7 The Fe atom exchange rates between Fe(Ⅱ)aq and structural Fe(Ⅲ) of ferrihydrite during the Fe(Ⅱ)-catalyzing transformation of ferrihydrite with or without coexistence of Pb(Ⅱ)
-


 下载:
下载:







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 28
- 文章访问数: 3326
- HTML全文浏览量: 622
 下载:
下载:
