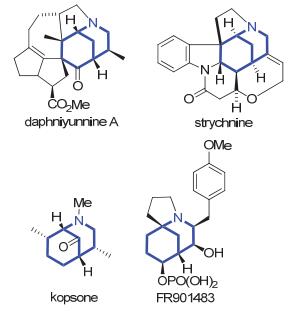
 图 1
含氮杂双环[3.3.1]壬烷结构的天然产物
Figure 1.
Representative natural products containing morphan azabicyclic nucleus
图 1
含氮杂双环[3.3.1]壬烷结构的天然产物
Figure 1.
Representative natural products containing morphan azabicyclic nucleus
引用本文:
李保乐, 刘人荣, 梁仁校, 贾义霞. 钯/氨基酸共催化环己酮分子内α-烯基化反应[J]. 化学学报,
2017, 75(5): 448-452.
doi:
10.6023/A17020080

Citation: Li Baole, Liu Renrong, Liang Renxiao, Jia Yixia. Palladium/Amino Acid Co-Catalyzed Intramolecular α-Vinylation of Cyclohexanones[J]. Acta Chimica Sinica, 2017, 75(5): 448-452. doi: 10.6023/A17020080
Citation: Li Baole, Liu Renrong, Liang Renxiao, Jia Yixia. Palladium/Amino Acid Co-Catalyzed Intramolecular α-Vinylation of Cyclohexanones[J]. Acta Chimica Sinica, 2017, 75(5): 448-452. doi: 10.6023/A17020080
钯/氨基酸共催化环己酮分子内α-烯基化反应
English
Palladium/Amino Acid Co-Catalyzed Intramolecular α-Vinylation of Cyclohexanones
Abstract:
Transition-metal-catalyzed α-vinylation of carbonyl compounds represents one of the most important carbon-carbon bond forming approaches to the synthesis of β, γ-unsaturated ketones, which are versatile synthetic building blocks and key structural motifs appearing in biologically active molecules. For this purpose, a number of methods have been developed by utilizing palladium-catalyzed cross-coupling of vinyl halide with C—H bond α-to carbonyl group. However, the elimination of vinyl halides in the presence of strong base would afford alkynes, which remained inert and therefore resulted in lower yields in the cross-coupling reaction. In recent years, cooperative catalysis merging transition metals and organic molecules represents a powerful strategy and renders numerous efficient transformations successful. Among which, palladium/enamine catalysis has emerged as an efficient method for the direct α-functionalization of ketones or aldehydes. We therefore envisaged that a direct cross-coupling of ketones and vinyl halides in the presence of Pd(0)/amine co-catalyst; the need of weak base would avoid the formation of alkynes through elimination of vinyl halides. Herein, we report a palladium/chiral amino acid co-catalyzed intramolecular α-vinylation reaction of cyclohexanones, which delivers a series of bridged ring compounds under mild reaction conditions in good to excellent yields. The resulting unique bridged ring system is analogous to the important morphan scaffold (2-azabicyclo[3.3.1]nonane), which is core structure existing in many important bioactive natural products. In the meantime, asymmetric version of this reaction was also tested and a number of desired products were achieved in moderate enantioselectivities. A representative procedure for this reaction is as following: To a dried Schlenk tube were added compound 1 (0.2 mmol), chiral amines (0.04 mmol, 20 mol%), K3PO4 (0.3 mmol, 1.5 equiv.), Pd(OAc)2 (0.01 mmol, 5 mol%) and PPh3 (0.024 mmol, 12 mol%) under N2, 2.0 mL THF was then introduced via a syringe. The resulting mixture was stirred at 85 ℃ (oil bath) for 72 h until the reaction was complete (monitored by TLC). The solvent was then removed under vacuum and the residue was purified by flash chromatography on silica gel, eluting with ethyl acetate/petroleum ether 1:10 (V/V) to afford the desired product.
-
Key words:
- palladium
- / amino acid
- / enamine
- / cyclohexanone
- / vinylation
-
1 引言
过渡金属催化羰基化合物α-烯基化反应是构建Csp3—Csp2键的重要方法之一, 近年来受到有机合成化学家的广泛关注.其中酮类化合物的α-烯基化反应获得的产物β, γ-不饱和酮是合成天然产物及药物分子的重要中间体[1]. 2001年, Buchwald小组[2]利用Pd2(dba)3与手性膦配体的络合物为催化剂, 通过取代环戊酮或环己酮的C—H键与烯基卤化物的交叉偶联反应, 高收率和对映选择性地实现了环酮的分子间α-烯基化反应. 2007年, Huang等[3]以[PdP(t-Bu)3Br]2为催化剂实现了2-吲哚酮及苯并环酮类化合物与烯基卤化物的α-烯基化反应.此外, 2010年Fu小组[4]利用α-卤代酮与烯基锆试剂的交叉偶联反应, 以NiCl2•glyme为催化剂手性双噁唑啉为配体, 高收率和高对映选择性地合成了一系列β, γ-不饱和酮类化合物.
2-氮杂双环[3.3.1]壬烷结构片段广泛存在于天然产物中, 是很多生物碱的核心结构(图 1)[5]. 2003年, Bonjoch小组[6]以Pd(PPh3)4为催化剂, 在叔丁醇钾作为碱的条件下实现了含有烯基卤化物片段的环己酮类化合物的分子内烯基化反应, 获得了氮杂双环[3.3.1]桥环化合物.然而可能由于烯基卤化物在强碱条件下很容易发生消除反应得到炔烃类副产物[7], 因而目标产物的收率较低.近年来, 过渡金属催化剂与有机小分子的共催化策略引起了化学家的持续关注, 并得到了深入研究和广泛应用, 使有机合成反应方法更加多样化[8].其中, 钯/烯胺共催化策略已被应用于不对称烯丙基烷基化反应[9]. 2016年, Dong等[10]报道了钯/四氢吡咯共催化分子间环戊酮α-芳基化反应.与此同时, 我们小组[11]成功利用钯/ L-脯氨酸共催化剂实现了环己酮分子内不对称芳基化反应, 高对映选择性地合成了一系列具有两个手性中心的桥环化合物.初步机理探索表明反应中脯氨酸和钯分别活化酮和芳基溴化物, 通过酮与氨基酸缩合得到的烯胺中间体的Heck反应得到产物, 因而只需弱碱的参与即能促进反应的发生.在此研究基础上, 我们进一步尝试环己酮分子内α-烯基化反应.这里我们报道该反应的初步结果.
图 1
含氮杂双环[3.3.1]壬烷结构的天然产物
Figure 1.
Representative natural products containing morphan azabicyclic nucleus
2 结果与讨论
2.1 反应条件优化
以化合物1a为模型底物, 我们研究分子内环己酮α-烯基化反应, 实验结果见表 1.首先, 当以5 mol% Pd(OAc)2为催化剂, 12 mol% PPh3为配体, 1.5 equiv. K3PO4为碱时, 在四氢呋喃溶剂中85 ℃下反应72 h后, 并没有获得目标产物2a(表 1, Entry 1).随后发现加入20 mol% L-脯氨酸A1为共催化剂时, 反应可观察到微量产物的生成(Entry 2).受此鼓舞, 我们在该反应中尝试了不同的氨基酸催化剂(Entries 3~7).当以L-苯丙氨酸A2为催化剂时, 可以42%的分离收率获得目标产物2a (Entry 3).以L-亮氨酸A3或L-缬氨酸A4为催化剂时, 产物2a的收率进一步提高至72%和78% (Entries 4~5).随后, 我们尝试了位阻更大的L-叔亮氨酸A5, 然而反应收率没有进一步明显提高, 最终以80%的收率分离得到目标产物2a (Entry 6).作为对照, 使用L-缬氨酸A4的酯化物A6导致反应收率严重降低至15% (Entry 7).在以较优的氨基酸A5为催化剂下, 我们考察了不同的碱对反应的影响.发现使用NaHCO3、CH3CO2Na及有机碱NEt3时反应不能发生(Entries 8, 9及12).而以K2CO3和Cs2CO3为碱时, 目标产物的收率分别降低至30%和48% (Entries 10~11).随后的溶剂实验表明反应在甲苯及甲醇中目标产物的收率极低(Entries 13~14).此外, 在乙腈中反应收率也仅获得43% (Entry 15).因此, 基于以上优化确定的较优反应条件为:以5 mol% Pd(OAc)2, 12% PPh3及20% L-叔亮氨酸为催化剂, 1.5 equiv. K3PO4为碱, 在四氢呋喃溶剂中, 85 ℃条件下反应.值得一提的是, 在所有反应条件下目标产物2a经手性HPLC检测均为外消旋.
 表 1
反应条件的优化a
Table 1.
Optimization of reaction conditions
表 1
反应条件的优化a
Table 1.
Optimization of reaction conditions

Entry Amine Base Solvent Yieldb/% 1 — K3PO4 THF ND 2 A1 K3PO4 THF <10 3 A2 K3PO4 THF 42 4 A3 K3PO4 THF 72 5 A4 K3PO4 THF 78 6 A5 K3PO4 THF 80 7 A6 K3PO4 THF 15 8 A5 NaHCO3 THF NR 9 A5 CH3COONa THF NR 10 A5 K2CO3 THF 30 11 A5 Cs2CO3 THF 48 12 A5 Et3N THF NR 13 A5 K3PO4 toluene <10 14 A5 K3PO4 MeOH 10 15 A5 K3PO4 MeCN 43 aReaction conditions: 1a (0.2 mmol), Pd(OAc)2 (5 mol%), PPh3 (12 mol%), amine (20 mol%), base (1.5 equiv.) in solvent (2.0 mL) at 85 ℃ for 72 h. bIsolated yields. 表 1 反应条件的优化a
Table 1. Optimization of reaction conditions2.2 底物拓展
在上述优化条件下, 我们考察了N原子的苯基上不同取代基对反应的影响(表 2).结果表明, 苯环的对位或间位上带有卤素取代基时, 反应均能以中等至良好的收率获得目标产物2c~2f.需要指出的是, 对氟苯基产物2e的收率较低, 而间氟苯基产物存在互变异构体2c2.在苯基上引入甲氧基时, 仅间甲氧基取代产物2b获得72%的收率, 而邻位或对位甲氧基取代底物的反应没有成功.其中邻甲氧基苯基底物的反应以75%的收率分离得到脱烯丙基产物3, 而对甲氧基苯基底物的反应结果较复杂, 未能分离获得任何纯净产物.此外, 苯环上对甲基取代底物的反应可获得目标化合物, 但其不稳定, 极易变质.当在烯烃末端引入甲基时, 可以83%的收率获得目标产物2g.随后, 我们又考察了N-苄基取代底物的反应, 反应在100 ℃下以46%的收率获得目标化合物2h, 然而令人高兴的是该产物经检测具有11%的对映选择性.同时发现烯烃末端引入顺式苯基时, 可以90%的收率获得目标产物2i, 其对映选择性提高至44%.进一步通过对底物结构的调整, 实现了含氮杂双环[3.4.1]桥环化合物的构建, 分别以65%和54%的收率获得目标化合物2j和2k.值得说明的是, 以氧原子为连接基团的底物在该优化条件下反应未能分离获得目标化合物2l.
表 2
分子内环己酮的α-烯基化反应a
Table 2.
Intramolecular α-vinylation of cyclohexanones


aReactions were carried out using 1a (0.2 mmol), Pd(OAc)2 (5 mol%), PPh3 (12 mol%), A5 (20 mol%), K3PO4 (1.5 equiv.), THF (2.0 mL), 85 ℃, 72 h; Isolated yields.b At 100 ℃ for 48 h. cAt 100 ℃ for 72 h. 表 2 分子内环己酮的α-烯基化反应a
Table 2. Intramolecular α-vinylation of cyclohexanones2.3 对映选择性反应研究
受上述对映选择性反应结果的鼓舞, 我们以底物1i的反应为模型优化了该环己酮去对称化的不对称α-烯基化反应.如表 3所示, 反应在手性氨基酸A2~A5促进下获得优异的收率(Entries 1~4), 其中L-缬氨酸A4可获得最高47%的对映选择性(Entry 3).对氟苯甘氨酸A7参与的反应收率有明显降低, 并只获得20%的对映选择性(Entry 5).而L-缬氨醇A8不能促进反应的发生(Entry 6).此外, 我们发现膦配体对反应有较大的影响.尽管PCy3和PtBu3配体能有效地促进反应, 但目标化合物2i的对映选择性极低(Entries 7~8),其中PCy3配体的反应几乎消旋(Entry 8).而亚磷酸三乙酯为配体时收率和对映选择性均明显降低(Entry 9).此外, 手性双膦配体BINAP能促进反应获得中等收率, 但产物几乎消旋(Entries 11~12).
表 3
环己酮去对称化不对称α-烯基化反应条件优化a
Table 3.
Condition optimization of enantioselective α-vinylation of cyclohexanones

Entry Amine L Yieldb/% eec/% 1 A2 PPh3 90 20 2 A3 PPh3 84 20 3 A4 PPh3 92 47 4 A5 PPh3 90 44 5 A7 PPh3 73 20 6d A8 PPh3 ND — 7 A4 PCy3 87 rac 8 A4 PtBu3 67 <10 9 A4 P(OEt)3 30 15 10 A4 P(p-Tol)3 78 40 11 A4 (S)-BINAP 58 <10 12 A4 (R)-BINAP 60 rac aReaction conditions: 1i (0.2 mmol), Pd(OAc)2 (5 mol%), L (12 mol%), amine (20 mol%), and K3PO4 (1.5 equiv.) in THF (2.0 mL) at 100 ℃ for 48 h. bIsolated yields. c Determined by chiral HPLC. d 20 mol% benzoic acid was added. 表 3 环己酮去对称化不对称α-烯基化反应条件优化a
Table 3. Condition optimization of enantioselective α-vinylation of cyclohexanones在上述较优条件下, 我们对不对称α-烯基化反应进行了底物拓展.首先, 当烯烃上苯基被甲基取代后, 目标产物2m的收率和对映选择性均明显降低(70%的收率和14%的ee值).其次, 考察了氮原子上的烷基取代基对反应的影响.由表 4可以发现, 反应都能以优良的收率获得相应产物2i和2n~2r. N-对氯苄基取代基以及N-苯乙基取代产物2n和2o的对映选择性均达到51%.此外, 发现氮原子上取代基的位阻效应对反应的对映选择性有一定的影响.位阻较小的甲基取代基的产物2p获得最好的对映选择性, 达到57%, 而位阻较大的异丙基取代产物2q的ee值则降为30%. N-正丁基取代产物2r与N-苄基产物2i的对映选择性相当.随后, 以N-甲基为保护基考察了苯基上的取代基效应.发现苯环带有甲基或卤素基团的底物在反应中均能以良好到优异的收率以及中等程度的对映选择性获得相应产物2s~2v.烯烃的立体构型对该反应有较大的影响.我们合成了反式苯基取代的烯烃底物1w, 遗憾地发现该底物在优化条件下并不发生反应, 未能获得目标产物2w.同样, 双苯基取代烯烃底物1x合成产物2x的反应也不发生.
表 4
不对称α-烯基化反应a
Table 4.
Asymmetric α-vinylation of cyclohexanones


aReaction conditions: 1 (0.2 mmol), Pd(OAc)2 (5 mol%), PPh3 (12 mol%), A4 (20 mol%), and K3PO4 (1.5 equiv.) in THF (2.0 mL) at 100 ℃ for 48 h; isolated yields and the ee was determined by chiral HPLC. 表 4 不对称α-烯基化反应a
Table 4. Asymmetric α-vinylation of cyclohexanones2.4 可能的反应机理
基于文献研究[11], 我们提出了如下可能的反应机理.如图式1所示, 手性氨基酸与环己酮的羰基作用生成烯胺中间体A.同时, 零价钯催化剂与碳溴键发生氧化加成反应获得烯基钯中间体B, 随后该烯基钯对烯胺发生插入反应得到中间体C, 再经β-氢消除后得到烯胺中间体D, 同时通过还原消除反应再生Pd(0) 催化剂.中间体D随后经水解释放氨基酸催化剂, 并获得目标产物2a.
 图 图式1
可能的反应机理
Figure 图式1.
Proposed mechanism for α-vinylation of cyclohexanones
图 图式1
可能的反应机理
Figure 图式1.
Proposed mechanism for α-vinylation of cyclohexanones
3 结论
以醋酸钯和简单易得的氨基酸作催化剂, 成功实现了分子内环己酮α-烯基化反应.在温和的反应条件下, 以中等到良好的收率合成了一系列桥环化合物, 并对不对称催化反应进行了初步尝试, 取得中等水平的对映选择性.反应以廉价易得的氨基酸活化环己酮, 在弱碱条件下实现α-烯基化反应, 为这类反应的研究提供了一种新的催化策略.
4 实验部分
分子内环己酮α-烯基化反应的一般操作步骤如下:在安培管中依次加入0.2 mmol的底物1, 5 mol%醋酸钯, 12 mmol%三苯基膦以及20 mol%氨基酸和1.5 equiv.磷酸钾.在氮气下置换三次后, 用注射器加入2 mL的新蒸馏的四氢呋喃溶剂, 在85 ℃或100 ℃下反应72 h或48 h.反应结束后粗产品用层析柱分离得到目标产物2a~2m [淋洗液: V(乙酸乙酯):V(石油醚, 60~90 ℃)=1:10].
-
-
[1]
(a) Ohtsuka, Y. ; Sasahara, T. ; Oishi, T. Chem. Pharm. Bull. 1982, 30, 1106 and references therein. (b) Gohain, M. ; Gogoi, B. J. ; Prajapati, D. ; Sandhu, J. S. New J. Chem. 2003, 27, 1038. (c) Shi, D. -Q. ; Zhang, S. ; Zhuang, Q. -Y. ; Tu, S. -J. ; Hu, H. -W. Chin. J. Org. Chem. 2003, 23, 1036 (in Chinese). (史达清, 张姝, 庄启亚, 屠树江, 胡宏纹, 有机化学, 2003, 23, 1036. ) (d) Comprehensive Organic Functional Group Transformations Ⅱ, Eds. : Katritzky, A. R. ; Taylor, R. J. K. , Elsevier, Amsterdam, 2005. (e) Iwasaki, M. ; Morita, E. ; Uemura, M. ; Yorimitsu, H. ; Oshima, K. Synlett 2007, 2007, 167. (f) Zhou, G. ; Yu, L. ; Hui, Y. ; Xie, Z. Acta Chim. Sinica 2012, 70, 1289 (in Chinese). (周广鹏, 余蕾, 惠永海, 解正峰, 化学学报, 2012, 70, 1289. ) (g) Liu, Y. ; Zhou, J. Acta Chim. Sinica 2012, 70, 1451. (in Chinese). (刘运林, 周剑, 化学学报, 2012, 70, 1451. )
-
[2]
Chieffi, A.; Kamikawa, K.; Ahman, J.; Fox, J. M.; Buchwald, S. L. Org. Lett. 2001, 3, 1897. doi: 10.1021/ol0159470
-
[3]
Huang, J.; Bunel, E.; Faul, M. M. Org. Lett. 2007, 9, 4343. doi: 10.1021/ol7019839
-
[4]
Lou, S.; Fu, G. C. J. Am. Chem. Soc. 2010, 132, 5010. doi: 10.1021/ja1017046
-
[5]
(a) Staub, G. M.; Gloer, J. B.; Wicklow, D. T.; Dowd, P. F. J. Am. Chem. Soc. 1992, 114, 1015. (b) Kan-Fan, C.; Sevenet, T.; Had, H. A.; Bonin, M.; Quirion, J.-C.; Husson, H.-P. Nat. Prod. Lett. 1996, 7, 283. (c) Kong, F.; Graziani, E. I.; Andersen, R. J. J. Nat. Prod. 1998, 61, 267. (d) Heathcock, C. H.; Clasby, M.; Griffith, D. A.; Henke, B. R.; Sharp, M. J. Synlett 1995, 1995, 467. (e) Takayama, H.; Katakawa, K.; Kitajima, M.; Seki, H.; Yamaguchi, K.; Aimi, N. Org. Lett. 2002, 4, 1243. (f) Bonjoch, J.; Diaba, F. Stud. Nat. Prod. Chem. 2005, 32, 3. (g) Magnus, P.; Padilla, A. I. Org. Lett. 2006, 8, 3569. (h) Kobayashi, J.; Kubota, T. Nat. Prod. Rep. 2009, 26, 936. (i) Rinner, U.; Lentsch, C.; Aichinger, C. Synthesis 2010, 2010, 3763. (j) Mori, M. Heterocycles 2010, 81, 259.
-
[6]
Solé, D.; Diaba, F.; Bonjoch, J. J. Org. Chem. 2003, 5746.
-
[7]
Piers, E.; Marais, P. C. J. Org. Chem. 1990, 55, 3454. doi: 10.1021/jo00298a014
-
[8]
(a) Shao, Z.; Zhang, H. Chem. Soc. Rev. 2009, 38, 2745. (b) Zhong, C.; Shi, X. Eur. J. Org. Chem. 2010, 2999.
-
[9]
(a) Ibrahem, I.; Córdova, A. Angew. Chem., Int. Ed. 2006, 45, 1952. (b) Afewerki, S.; Ibrahem, I.; Rydfjord, J.; Breistein, P.; Córdova, A. Chem. Eur. J. 2012, 18, 2972. (c) Mukherjee, S.; List, B. J. Am. Chem. Soc. 2007, 129, 11336. (d) Zhao, X. H.; Liu, D. L.; Guo, H.; Liu, Y. G.; Zhang, W. B. J. Am. Chem. Soc. 2011, 133, 19354. (e) Wu, H.; He, Y. P.; Gong, L. Z. Adv. Synth. Catal. 2012, 354, 975. (f) Tao, Z. L.; Zhang, W. Q.; Chen, D. F.; Adele, A.; Gong, L. Z. J. Am. Chem. Soc. 2013, 135, 9255. (g) Krautwald, S.; Sarlah, D.; Schafroth, M. A.; Carreira, E. M. Science 2013, 340, 1065. (h) Yoshida, M.; Terumine, T.; Masaki, E.; Hara, S. J. Org. Chem. 2013, 78, 10853. (i) Krautwald, S.; Schafroth, M. A.; Sarlah, D.; Carreira, E. M. J. Am. Chem. Soc. 2014, 136, 3020. (j) Huo, X.; Yang, G.; Liu, D.; Liu, Y.; Gridnev, I. D.; Zhang, W. Angew. Chem., Int. Ed. 2014, 53, 6776. (k) Huo, X.; Quan, M.; Yang, G. Q.; Zhao, X. H.; Liu, D. L.; Liu, Y. G.; Zhang, W. B. Org. Lett. 2014, 16, 1570. (l) Tang, S.; Wu, X. D.; Liao, W. Q.; Liu, K.; Liu, C.; Luo, S. Z.; Lei, A. W. Org. Lett. 2014, 16, 3584. (m) Wang, P. S.; Lin, H. C.; Zhai, Y. J.; Han, Z. Y.; Gong, L. Z. Angew. Chem., Int. Ed. 2014, 53, 12218. (n) Zhou, X. L.; Wang, P. S.; Zhang, D. W.; Liu, P.; Wang, C. M.; Gong, L. Z. Org. Lett. 2015, 17, 5120. (o) Zhou, H.; Zhang, L.; Xu, C.; Luo, S. Angew. Chem., Int. Ed. 2015, 54, 12645.
-
[10]
Xu, Y.; Su, T.; Huang, Z.; Dong, G. Angew. Chem., Int. Ed. 2016, 55, 2559. doi: 10.1002/anie.201510638
-
[11]
Liu, R.-R.; Li, B.-L.; Lu, J.; Shen, C.; Gao, J.-R.; Jia, Y.-X. J. Am. Chem. Soc. 2016, 138, 5198. doi: 10.1021/jacs.6b01214
-
[1]
-

图 1 含氮杂双环[3.3.1]壬烷结构的天然产物
Figure 1 Representative natural products containing morphan azabicyclic nucleus
表 1 反应条件的优化a
Table 1. Optimization of reaction conditions
Entry Amine Base Solvent Yieldb/% 1 — K3PO4 THF ND 2 A1 K3PO4 THF <10 3 A2 K3PO4 THF 42 4 A3 K3PO4 THF 72 5 A4 K3PO4 THF 78 6 A5 K3PO4 THF 80 7 A6 K3PO4 THF 15 8 A5 NaHCO3 THF NR 9 A5 CH3COONa THF NR 10 A5 K2CO3 THF 30 11 A5 Cs2CO3 THF 48 12 A5 Et3N THF NR 13 A5 K3PO4 toluene <10 14 A5 K3PO4 MeOH 10 15 A5 K3PO4 MeCN 43 aReaction conditions: 1a (0.2 mmol), Pd(OAc)2 (5 mol%), PPh3 (12 mol%), amine (20 mol%), base (1.5 equiv.) in solvent (2.0 mL) at 85 ℃ for 72 h. bIsolated yields.  下载: 导出CSV
下载: 导出CSV
表 2 分子内环己酮的α-烯基化反应a
Table 2. Intramolecular α-vinylation of cyclohexanones
aReactions were carried out using 1a (0.2 mmol), Pd(OAc)2 (5 mol%), PPh3 (12 mol%), A5 (20 mol%), K3PO4 (1.5 equiv.), THF (2.0 mL), 85 ℃, 72 h; Isolated yields.b At 100 ℃ for 48 h. cAt 100 ℃ for 72 h.
下载: 导出CSV
表 3 环己酮去对称化不对称α-烯基化反应条件优化a
Table 3. Condition optimization of enantioselective α-vinylation of cyclohexanones
Entry Amine L Yieldb/% eec/% 1 A2 PPh3 90 20 2 A3 PPh3 84 20 3 A4 PPh3 92 47 4 A5 PPh3 90 44 5 A7 PPh3 73 20 6d A8 PPh3 ND — 7 A4 PCy3 87 rac 8 A4 PtBu3 67 <10 9 A4 P(OEt)3 30 15 10 A4 P(p-Tol)3 78 40 11 A4 (S)-BINAP 58 <10 12 A4 (R)-BINAP 60 rac aReaction conditions: 1i (0.2 mmol), Pd(OAc)2 (5 mol%), L (12 mol%), amine (20 mol%), and K3PO4 (1.5 equiv.) in THF (2.0 mL) at 100 ℃ for 48 h. bIsolated yields. c Determined by chiral HPLC. d 20 mol% benzoic acid was added.
下载: 导出CSV
表 4 不对称α-烯基化反应a
Table 4. Asymmetric α-vinylation of cyclohexanones
aReaction conditions: 1 (0.2 mmol), Pd(OAc)2 (5 mol%), PPh3 (12 mol%), A4 (20 mol%), and K3PO4 (1.5 equiv.) in THF (2.0 mL) at 100 ℃ for 48 h; isolated yields and the ee was determined by chiral HPLC.
下载: 导出CSV
-



 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 27
- 文章访问数: 2593
- HTML全文浏览量: 375
 下载:
下载:
