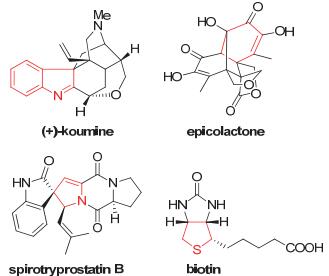
 图 1
天然产物中芳香化合物骨架
Figure 1.
Natural products containing aromatic compound skeleton
图 1
天然产物中芳香化合物骨架
Figure 1.
Natural products containing aromatic compound skeleton
引用本文:
吴文挺, 张立明, 游书力. 金催化去芳构化反应研究进展[J]. 化学学报,
2017, 75(5): 419-438.
doi:
10.6023/A17020049

Citation: Wu Wen-Ting, Zhang Liming, You Shu-Li. Recent Progress on Gold-catalyzed Dearomatization Reactions[J]. Acta Chimica Sinica, 2017, 75(5): 419-438. doi: 10.6023/A17020049
Citation: Wu Wen-Ting, Zhang Liming, You Shu-Li. Recent Progress on Gold-catalyzed Dearomatization Reactions[J]. Acta Chimica Sinica, 2017, 75(5): 419-438. doi: 10.6023/A17020049
金催化去芳构化反应研究进展
English
Recent Progress on Gold-catalyzed Dearomatization Reactions
Abstract:
Homogeneous gold catalysis has experienced rapid development since 2004 and generally exhibited high efficiency and good functional group tolerance. On the other hand, catalytic dearomatization reactions provide a unique and straight approach to the construction of highly functionalized molecules with diverse three-dimensional structures from simple aromatic compounds. In this perspective, recent examples on gold-catalyzed dearomatization reactions are summarized in two main categories: gold-catalyzed rearrangements and gold-catalyzed hydrofunctionalizations of alkynes and allenes. In the first category, intra-and inter-molecular dearomatization reactions were achieved via gold-catalyzed rearrangements of propargylic ester and its derivatives. Although this area is still at its early stage, several outstanding asymmetric examples have been reported by Shi and Toste. In the second category, an array of dearomatization reactions via gold-catalyzed hydrofunctionalizations of alkynes and allenes were presented. All these cases have shown great potentials for convenient and straightforward construction of spiro and/or bridged polycyclic molecules, and some of them have exhibited excellent enantioselectivity. In addition, salient features and proposed mechanisms for these two types of reactions are also described.
-
Key words:
- gold catalysis
- / dearomatization
- / asymmetric catalysis
- / rearrangement
- / hydrofunctionalization
-
1 引言
长期以来, 金一直被认为是没有催化活性的贵金属.直到1986年Ito和Hayashi等[1]将Au(Ⅰ)络合物应用于催化不对称aldol反应中, 这样的观念才被打破. 2000年前后, Au(Ⅰ)催化剂被发现能够非常高效地活化炔烃从而发生炔烃的氢-官能团化反应[2], 自此金催化发展迅速, 成为了一个十分热门的领域, 特别是均相金催化领域涌现出了许多引人注目的成果[3, 4].金催化剂凭借其独特的亲电性能够活化炔烃, 这已经成为均相金催化为人所熟知的特点, 并且金催化剂在天然产物或复杂有机分子的合成转化中得到了广泛的应用, 展现出了非常高的催化效率和非常强的官能团兼容性[5].
均相金催化目前研究主要集中于Au(Ⅰ)和Au(Ⅲ)作为软亲碳Lewis酸通过配位来活化C—C不饱和键.高亲碳性也说明金的亲氧性相对较弱, 能够形成较为稳定的M—C键, 从而保证了催化剂的高效循环.此外, Au(Ⅰ)和Au(Ⅲ)之间的氧化电势很高, 这使得金催化剂对空气不敏感.因此, 相较于其他过渡金属催化剂, 金络合物可以在十分温和的条件下催化反应(可以容忍空气和水汽), 具有高活性、高化学选择性以及高官能团容忍性.
金阳离子由于其特殊的电子结构[6]而具有很强的π-酸性, 能够有效活化烯烃、联烯和炔烃, 发生氢-官能团化反应, 环加成反应, 炔丙醇类化合物重排反应, aldol反应, 炔烃参与的氧原子转移反应, 烯炔环化反应等[7].
另一方面, 催化去芳构化反应[8]能够直接高效地将平面的、相对易得的芳香化合物转化为高度官能团化的具有丰富三维立体结构的分子, 快速构建季碳中心以及桥环和并环结构.并且, 去芳构化的产物结构在众多天然产物以及具有生物活性分子中广泛存在.因此, 利用去芳构化策略来发展快速高效的合成路线具有重要的意义(Figure 1).
图 1
天然产物中芳香化合物骨架
Figure 1.
Natural products containing aromatic compound skeleton
众所周知, 金催化常被用于芳环以及芳杂环的合成[9]和衍生化[10].而将金催化应用于去芳构化反应中, 将会为天然产物和复杂分子的合成提供新颖直接高效的路径.虽然目前报道相对有限, 但已经展现出了非常大的发展潜力.
本文将根据金催化反应类型来介绍金催化去芳构化反应的研究进展.目前, 文献报道的金催化去芳构化反应主要集中于金催化的重排反应和炔烃/联烯的氢-官能团化反应, 以下将分这两方面进行介绍.
2 通过重排反应实现的金催化去芳构化反应
在过去均相金催化蓬勃发展的十几年间, 金催化的炔丙酯及其衍生物的重排反应吸引了众多研究者的兴趣.其中炔丙酯类底物用的较多, 它们的金催化重排反应通常可经过如下两个路径: 1, 2-迁移或3, 3-迁移, 分别形成金卡宾中间体和金配位的联烯中间体(Scheme 1), 这些中间体可以发生进一步的转化.通常认为影响反应最终产物的因素有:炔烃上的取代基团, 与炔丙酯直接相连的取代基团的电性, 迁移基团的性质, 催化剂的Lewis酸性, 底物中是否含有Lewis碱性或亲核性的位点等.
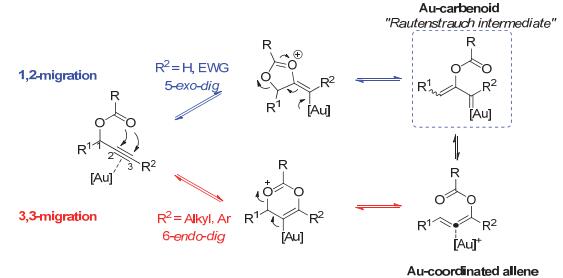 图 图式1
金催化的炔丙酯及其衍生物的重排反应的两条可能反应路径
Figure 图式1.
Two pathways for gold-catalyzed rearrangements of propargyl ester and its derivatives
图 图式1
金催化的炔丙酯及其衍生物的重排反应的两条可能反应路径
Figure 图式1.
Two pathways for gold-catalyzed rearrangements of propargyl ester and its derivatives
2.1 分子内反应
2005年, 张立明小组[11]报道了首例通过金催化炔丙酯[3, 3]-迁移重排反应串联[2+2]环加成实现的对吲哚类底物的去芳构化反应.在金催化条件下, 吲哚衍生的炔丙酯类化合物1可以在室温下高效地转化为2, 3-吲哚啉并四元环产物2, 收率最高可达98%.作者推测, 在反应中, 金催化剂首先活化C≡C从而发生3-吲哚乙酰氧基的[3, 3]-迁移重排, 形成被金催化剂配位的联烯中间体2-A, 随后形成氧鎓离子中间体2-B和2-B'.由于C(sp2)-Au较长, 所以[Au]与R1处在cis位置的中间体2-B所受张力更小, 热力学上更为稳定. 2-B中吲哚3位亲核进攻氧鎓离子环化形成中间体2-C, 最后分子内的烯基-金物种捕获产生的亚铵正离子生成含有E式环外双键的四元环产物2 (Scheme 2).
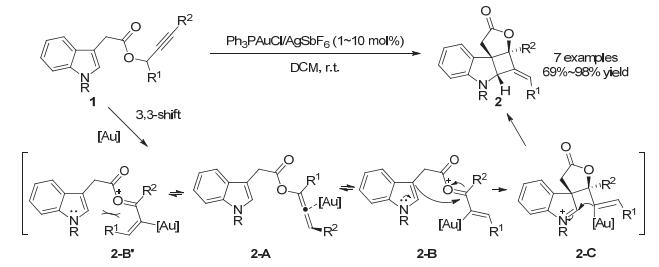 图 图式2
金催化炔丙酯[3, 3]-迁移重排[2+2]环加成串联反应
Figure 图式2.
Tandem Au-catalyzed 1, 3-rearrangement-[2+2] cycloadditions of propargylic esters
图 图式2
金催化炔丙酯[3, 3]-迁移重排[2+2]环加成串联反应
Figure 图式2.
Tandem Au-catalyzed 1, 3-rearrangement-[2+2] cycloadditions of propargylic esters
2016年, 施敏小组[12]改变了吲哚上边链连接位置和顺序, 实现了金催化炔丙酯[1, 2]-迁移重排串联[3+2]环加成反应.从含有吲哚N上连有炔丙酯边链的底物3出发, 选用不同的金催化剂, 可以分别高效地得到氧桥联多环吲哚啉4和开环多环吲哚啉5.并且产物4在[IPrAu(CH3CN)][SbF6]催化下可水解得到产物5.作者推测反应首先经历金催化的炔丙酯的1, 2-乙酰氧基迁移重排反应, 形成金卡宾中间体3-A.随后羰基亲核进攻金卡宾形成1, 3-偶极子, 最后分子内的[3+2]环加成反应在无水条件下生成氧桥联多环吲哚啉4, 或者有水存在下水解生成开环多环吲哚啉5 (Scheme 3A).接着作者尝试了该反应的不对称催化.由于一价金阳离子的线性配位模式, 使得不对称金催化非常具有挑战性.在筛选大量手性配体后, 作者发现Feringa亚磷酰胺配体为最优配体.在优化的条件下, 以中等到良好的手性控制(62%~90% ee)一步构建了连续的四个手性中心(Scheme 3B).
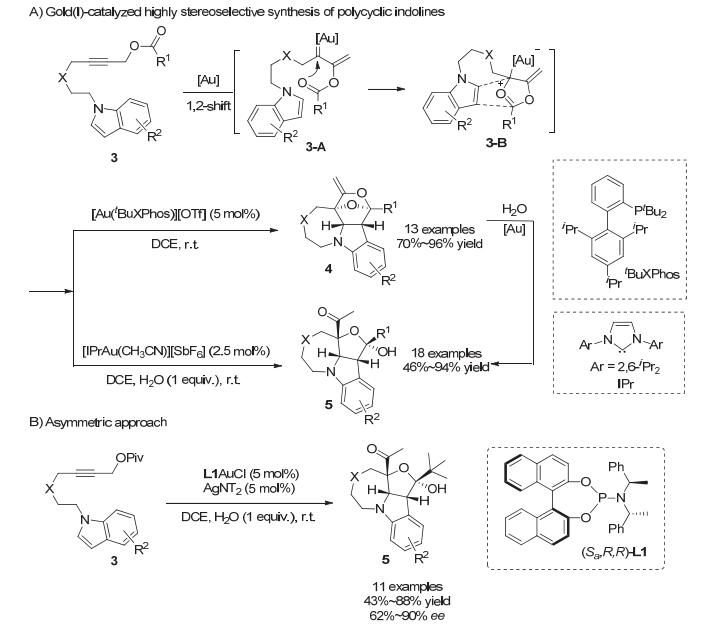 图 图式3
金催化炔丙酯[1, 2]-迁移重排[3+2]环加成串联反应
Figure 图式3.
Tandem Au-catalyzed 1, 2-rearrangement-[3+2] cycloadditions of propargylic esters
图 图式3
金催化炔丙酯[1, 2]-迁移重排[3+2]环加成串联反应
Figure 图式3.
Tandem Au-catalyzed 1, 2-rearrangement-[3+2] cycloadditions of propargylic esters
2015年Toste小组[13]报道了通过不对称金催化Rautenstrauch重排反应实现了对吲哚类底物的催化不对称去芳构化反应.将之前的炔丙酯底物换为炔丙基缩醛6时, 在手性金催化剂条件下以良好到优秀的对映选择性(71%~97% ee)得到手性环戊[b]并吲哚(cyclopenta[b]indoles)产物7.作者推测, 以6a为例, 底物中半缩醛上的乙氧基醚反式进攻被金活化的炔烃, 形成氧鎓离子物种4-A. 4-A中C—O键快速断裂形成非环状的中间体4-B, 随后释放出乙醛从而生成金取代的1-氨基戊二烯基(1-aminopentadienyl)中间体4-C(中间体4-C也可表示为其卡宾共振结构中间体4-C').随后的手性金催化剂控制的imino-Nazarov环化反应决定了反应的对映选择性, 最后水解得到手性产物7a.正是使用了半缩醛衍生的底物, 避免了使用炔丙酯类底物会形成的环状中间体4-A', 使得Rautenstrauch重排反应经历了一个平面的、非手性的中间体4-C, 也因此使得该环化反应为一个配体控制的过程而非手性传递过程(Scheme 4).
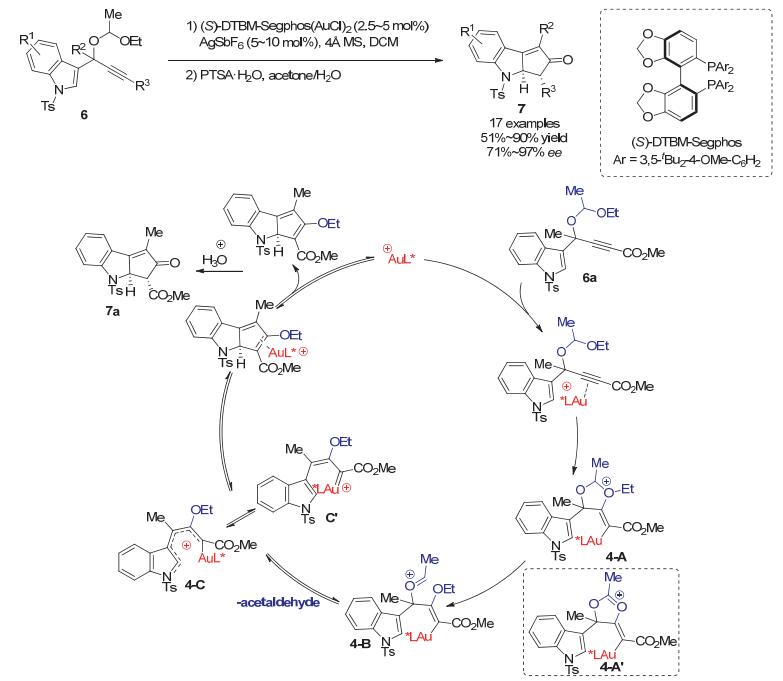 图 图式4
不对称金催化去芳构化Rautenstrauch重排反应
Figure 图式4.
Gold(Ⅰ)-catalyzed asymmetric dearomative Rautenstrauch rearrangement
图 图式4
不对称金催化去芳构化Rautenstrauch重排反应
Figure 图式4.
Gold(Ⅰ)-catalyzed asymmetric dearomative Rautenstrauch rearrangement
2.2 分子间反应
炔丙酯及其衍生物在金催化下发生重排反应而生成的中间体除了与分子内的活性位点发生反应外, 还能与另一活性分子发生反应, 迅速构建更为复杂的分子. 2008年, 张立明小组[14]报道了通过金催化重排反应生成的含金全碳1, 4-偶极体参与的[4+2]环化反应实现了吲哚分子间去芳构化反应, 能够快速高效得到多并环吲哚啉类产物10.作者推测1-(1-炔基)环丙基酮8在金催化条件下会发生重排反应, 经历氧鎓离子中间体5-A后环丙烷开环生成含金的全碳的1, 4-偶极体5-B.由于链长限制, 这样的电荷分离的中间体5-B并没有发生自身的环化反应, 而是与吲哚双键发生[4+2]环化反应, 从而以优秀的收率(83%~91%)得到多并环吲哚啉类产物10 (Scheme 5).
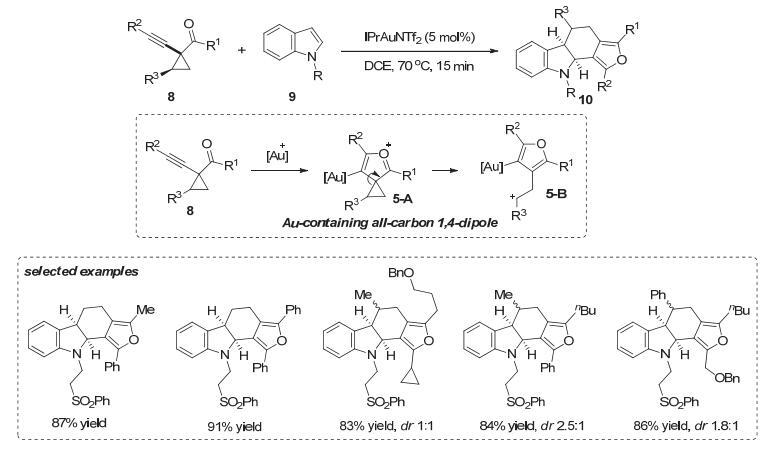 图 图式5
金催化重排反应生成的含金全碳1, 4-偶极体参与的[4+2]环化反应
Figure 图式5.
Au-Containing all-carbon 1, 4-dipoles: generation and [4+2] annulation
图 图式5
金催化重排反应生成的含金全碳1, 4-偶极体参与的[4+2]环化反应
Figure 图式5.
Au-Containing all-carbon 1, 4-dipoles: generation and [4+2] annulation
金催化重排反应除了可以产生含金的全碳的1, 4-偶极体以外, 还可以产生含金的全碳的1, 3-偶极体从而发生[3+2]环加成反应. 2008年, 张立明小组[15]报道了这样的偶极体的生成及其反应, 并且当另一分子底物为苄基保护的吲哚时, 实现了吲哚分子间去芳构化反应.反应过程中炔丙基半缩醛11在金催化下发生[1, 2]-迁移-乙醛脱除反应, 并存在更为稳定的共振形式含金1, 3-偶极体6-L.随后与吲哚双键发生[3+2]环加成反应, 以55%收率得到五元环并吲哚啉12 (Scheme 6).
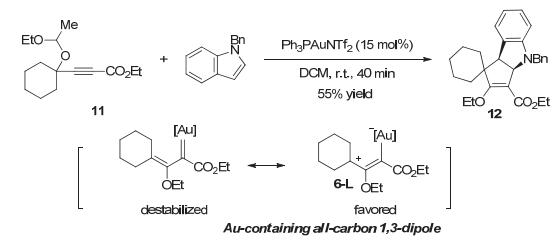 图 图式6
金催化重排反应生成的含金全碳1, 3-偶极体参与的[3+2]环化反应
Figure 图式6.
Au-containing all-carbon 1, 3-dipoles: generation and [3+2] cycloaddition reactions
图 图式6
金催化重排反应生成的含金全碳1, 3-偶极体参与的[3+2]环化反应
Figure 图式6.
Au-containing all-carbon 1, 3-dipoles: generation and [3+2] cycloaddition reactions
2012年, Davies小组[16]在研究金催化内炔的不对称环丙烯化反应时, 在底物拓展过程中意外发现, 化合物13与14反应并没有得到预期的环丙烯化的产物, 而是以83%的收率得到了五并七元环的双环产物15.作者推测金催化剂与重氮化合物原位生成的金卡宾物种接受炔烃的进攻生成阳离子物种7-A, 边链上的芳环随后进攻烯基阳离子形成双环中间体7-B, 中间体7-B经历重排形成含有环丙烷并环己二烯结构的中间体7-C, 最后环状双烯经历6π电开环反应生成环庚三烯衍生物15, 但是原本优秀的对映选择性下降至18% ee (Scheme 7).
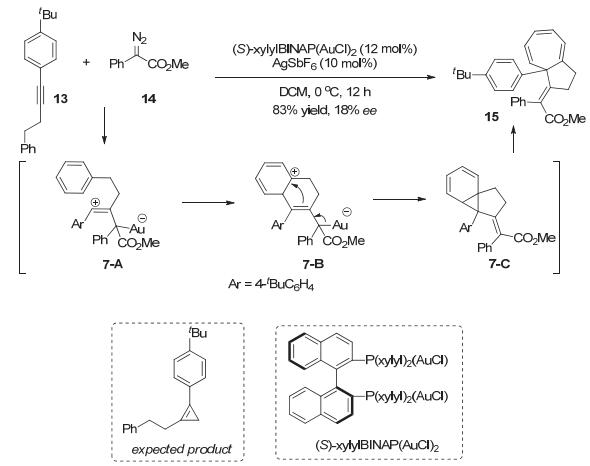 图 图式7
金催化内炔烃不对称环丙烯化反应研究中意外发现的重排反应
Figure 图式7.
Unexpected rearrangement in the study of gold(Ⅰ)-catalyzed asymmetric cyclopropenation of internal alkynes
图 图式7
金催化内炔烃不对称环丙烯化反应研究中意外发现的重排反应
Figure 图式7.
Unexpected rearrangement in the study of gold(Ⅰ)-catalyzed asymmetric cyclopropenation of internal alkynes
金卡宾物种除了可以由重氮化合物和金催化剂原位生成以外, 还可以在金催化炔基叠氮环化合成吲哚的过程中生成. 2014年, Fujii和Ohno小组[17]使用叠氮炔胺化合物16作为合成吲哚的底物, 发现在底物中如果含有双键, 那么合成吲哚过程中生成的中间体8-A中的金卡宾会与之发生环丙烷化, 最后以良好到优秀的收率生成多环假吲哚结构17 (Scheme 8).
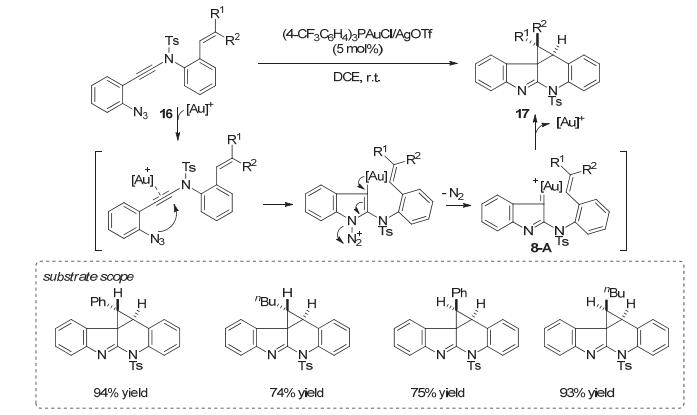 图 图式8
金催化叠氮炔胺的串联环化反应
Figure 图式8.
Gold-catalyzed cascade cyclization of (azido)ynamides
图 图式8
金催化叠氮炔胺的串联环化反应
Figure 图式8.
Gold-catalyzed cascade cyclization of (azido)ynamides
3 通过炔烃/联烯的氢-官能团化反应实现的金催化去芳构化反应
过渡金属催化不饱和C—C键的氢-官能团化是一类非常重要的反应[18], 分子内的反应常被用于构建含碳或含杂原子的环状分子骨架.由于金催化剂的高亲碳性, 能够通过配位来活化C—C不饱和键, 因此金催化在这一领域有非常多的应用.以炔烃为例, 金催化剂通过配位活化C≡C, 使其接受亲核试剂的反式进攻, 形成烯基金物种, 该中间体可以发生进一步转化, 生成稳定的产物.这里的亲核试剂可以是含O、N等杂原子, 也可以是含C的亲核试剂(Scheme 9A).例如使用含N亲核试剂的底物, 在金催化下可以用来合成吲哚(Scheme 9B).当亲核试剂为富电子的芳环时, 则该反应一般被称之为炔烃的芳基化反应, 也可以看做是傅克(Friedel-Crafts)反应类型的芳烃烯基化反应.例如, 当以吲哚为亲核试剂时, 则实现了金催化的炔烃氢-吲哚化(Scheme 9C).反应过程中涉及去芳构化的带正电荷的中间体(当芳环为吲哚时, 一般形成亚铵正离子), 为了得到稳定的产物, 一般会通过重新芳构化来实现, 得到芳环衍生化的产物.另一方面, 如果能够通过其他反应路径来得到稳定产物, 例如离去基团的离去或重排过程等, 分子内或分子间亲核试剂对高活性中间体进行捕获等, 则可以在产物中保留去芳构化的骨架, 从而实现金催化不饱和C—C键的氢-官能团化的去芳构化反应.下文将分成分子内反应和分子间反应进行介绍, 其中分子内反应将根据得到稳定产物的反应路径细分为通过离去基团的离去或重排反应实现的分子内环化直接去芳构化反应, 和通过分子内或分子间亲核试剂对高活性中间体进行捕获实现的分子内环化串联去芳构化反应.
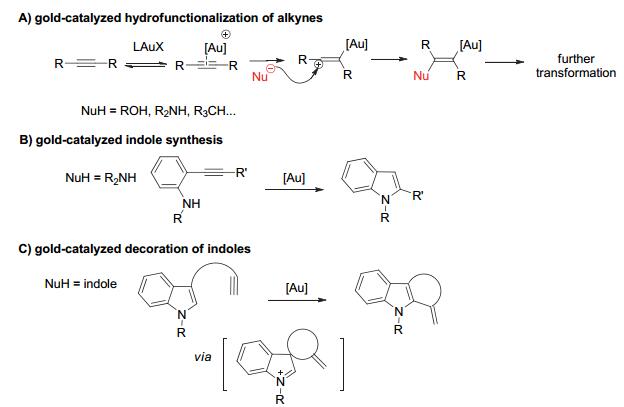 图 图式9
金催化炔烃的氢-官能团化反应模式及其在吲哚合成和修饰中的应用
Figure 图式9.
General model for gold-catalyzed hydrofunctionalization of alkynes and its application in the synthesis and decoration of indoles
图 图式9
金催化炔烃的氢-官能团化反应模式及其在吲哚合成和修饰中的应用
Figure 图式9.
General model for gold-catalyzed hydrofunctionalization of alkynes and its application in the synthesis and decoration of indoles
3.1 分子内环化直接去芳构化反应
在这一分类中, 大多数反应过程中都经历了金催化的环化反应形成含有金的带正电荷的去芳构化中间体, 为了得到稳定的产物, 在分子内没有亲核位点时, 通过离去基团的离去来生成稳定的产物, 一般得到螺环产物.文献中报道的形式上的离去基团有质子、酚氧基、甲基等.
2006年, Echavarren小组[19]在研究金催化的吲哚与炔烃的分子内反应时, 利用他们小组发展的Echavarren金催化剂Cat 1可以将带有炔基边链的底物18以65%的收率转化为七元环产物19, 并且该反应有较好的底物普适性[20], 作者推测反应是由吲哚3-位进攻被金配位活化的炔烃发生6-exo-dig环化反应生成含有金的亚铵正离子中间体10-A, 由于亚铵正离子相对不稳定, 随后[1, 2]-迁移重排芳构化后生成形式上吲哚傅克烯基化产物.当使用带有炔基边链的1, 2-(Me)2-吲哚底物20时, 可以80%的收率得到去芳构化的螺环产物21, 进一步支持了环化反应可能经历螺环中间体10-A的推测.该反应是第一例通过金催化炔烃的氢-官能团化反应实现的去芳构化反应, 通过脱去吲哚2-甲基上的氢形成环外双键从而得到稳定的产物21.同时, 作者还发现当端炔上的H被Me取代时, 会得到桥环产物23 (Scheme 10).作者推测底物22首先在金催化条件下发生7-endo-dig环化反应生成活性的亚铵正离子中间体10-B, 随后[1, 2]-迁移扩环得到中间体10-C, 接着失去质子得到中间体10-D, 在质子协助下发生八元环开环得到阳离子中间体10-E, 进而环化得到共轭的金卡宾中间体10-F, 随后发生迈克尔(Michael)类型的环化反应得到中间体10-G, 进一步发生质子化去金属化过程, 得到最终桥环产物23.
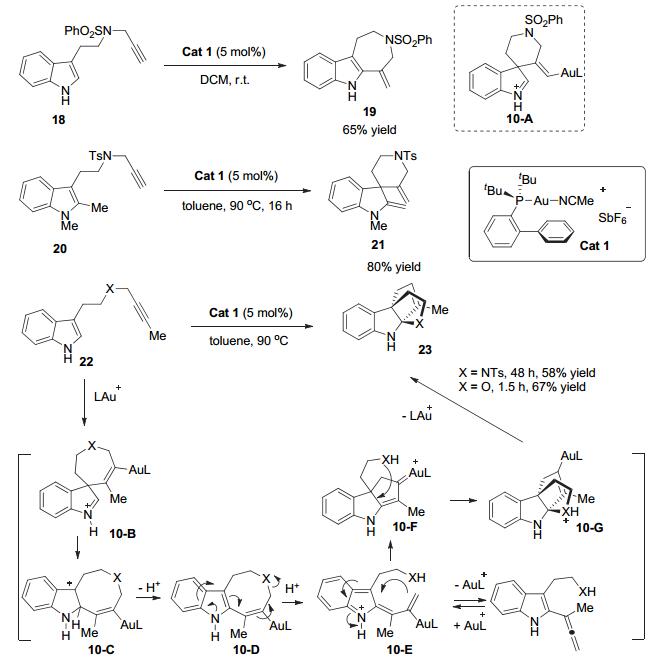 图 图式10
第一例通过金催化炔烃的氢-官能团化反应实现的去芳构化反应
Figure 图式10.
First example of dearomatization reaction via gold-catalyzed hydrofunctionalization of alkynes
图 图式10
第一例通过金催化炔烃的氢-官能团化反应实现的去芳构化反应
Figure 图式10.
First example of dearomatization reaction via gold-catalyzed hydrofunctionalization of alkynes
2011年, 涂永强小组[21]报道了吲哚C3位带有苯氧基取代的炔基吲哚24在金催化条件下也能发生环化反应, 当吲哚N上为吸电子取代基时, 底物会发生吲哚C2环化-苯氧基离去得到螺-假羟基吲哚类化合物25.同位素标记实验表明, C2环化去芳构产物中羰基O来源于溶剂中存在的水.进一步理论计算[22]详细阐释了反应机理.首先, 在一价金阳离子的催化下进行5-exo-dig环化生成带正电荷的中间体11-A, 随后在TfO-作为质子受体下被水捕获形成半缩酮结构11-B, 随后苯酚消除和烯基金部分质子化得到去芳构化产物25.在后续的苯酚消除和金属质子化过程中, 体系中生成的HOTf充当质子转移试剂, 对两者都有促进作用(Scheme 11).
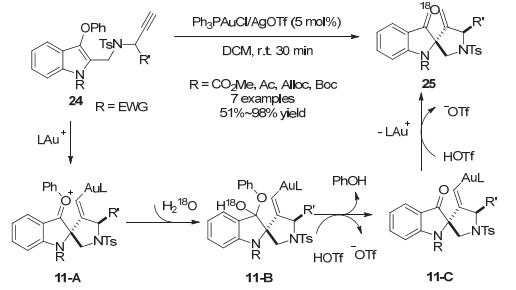 图 图式11
金催化吲哚C2位环化C3位苯酚氧基离去反应
Figure 图式11.
Gold-catalyzed indole C2-site annulation and removal of phenoxy group at the C3-site
图 图式11
金催化吲哚C2位环化C3位苯酚氧基离去反应
Figure 图式11.
Gold-catalyzed indole C2-site annulation and removal of phenoxy group at the C3-site
2015年, Wang小组[23]报道了通过金催化的脱硅基环化反应来合成桥环四环假吲哚化合物27, 这是在金催化环化反应中首例使用吲哚N-被硅基保护的底物.这里的硅基对于反应的选择性十分重要, 使反应选择性地发生C—C环化反应, 而不是C—N环化过程.从硅基保护的底物26出发, 反应经历了由吲哚C3位进攻被金活化炔烃而生成桥环中间体12-A, 在没有亲核试剂存在的条件下, 硅基离去并质子化去金化, 以中等到良好的收率(44%~86%)得到在天然产物中广泛存在的桥环四环假吲哚骨架27.在这里, 甲醇既作为硅基攫取剂, 也是质子源.最后, 作者对产物的分子活性进行了筛选, 发现其中一个产物能够选择性抑制MRSA(耐甲氧西林金黄色葡萄球菌)对β-内酰胺类抗生素的耐药性, 并有望发展成为针对具有耐药性细菌感染的辅助疗法(Scheme 12).
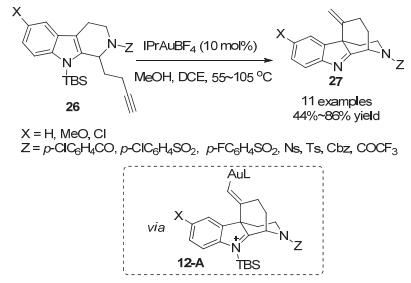 图 图式12
首例使用吲哚N硅基保护衍生物的金催化去芳构化环化反应
Figure 图式12.
First report of gold-catalyzed dearomative cyclization of N-silyl indole derivatives
图 图式12
首例使用吲哚N硅基保护衍生物的金催化去芳构化环化反应
Figure 图式12.
First report of gold-catalyzed dearomative cyclization of N-silyl indole derivatives
2016年, Fujii和Ohno小组[24]调整了底物中炔基边链长度并引入羟基, 合成了四氢咔唑衍生物28, 在金催化条件下发生6-endo-dig环化反应以76%收率生成桥环产物29, 完成了具有笼状结构的akuammiline生物碱(±)-strictamine的形式合成.值得指出的是, 反应中吲哚N—H不需要硅基保护(Scheme 13).
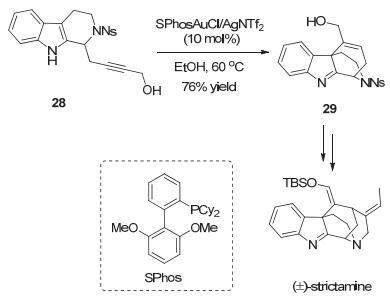 图 图式13
基于金催化环化反应的(±)-strictamine的形式全合成
Figure 图式13.
Formal total synthesis of (±)-strictamine based on a gold-catalyzed cyclization
图 图式13
基于金催化环化反应的(±)-strictamine的形式全合成
Figure 图式13.
Formal total synthesis of (±)-strictamine based on a gold-catalyzed cyclization
2015年, 王少仲和余志祥小组[25]在之前工作和理论计算的基础上, 使用2-炔丙基-β-四氢咔啉30作为底物, 在金催化剂和甲磺酸存在条件下, 当使用的炔烃为非端炔时, 发生碎裂-扩环反应, 以良好到优秀的收率得到中环产物azocinoindole衍生物31; 当使用的炔烃为端炔时, 发生螺环化反应, 以良好到优秀的收率主要得到螺环桥环产物32 (Scheme 14).
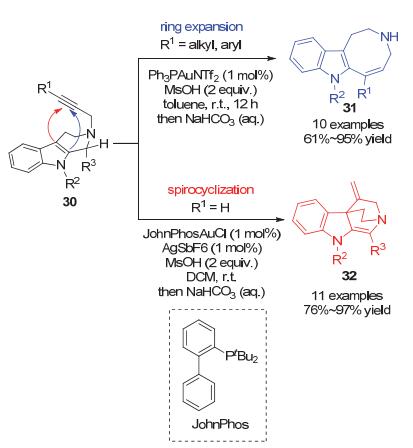 图 图式14
金催化2-炔丙基-β-四氢咔唑的扩环和螺环化反应
Figure 图式14.
Gold-catalyzed ring expansion and spirocyclization of 2-propargyl-β-tetrahydrocarbolines
图 图式14
金催化2-炔丙基-β-四氢咔唑的扩环和螺环化反应
Figure 图式14.
Gold-catalyzed ring expansion and spirocyclization of 2-propargyl-β-tetrahydrocarbolines
在金催化C—C不饱和键氢-官能团化反应中除了吲哚衍生物作为亲核试剂以外, 苯酚衍生物也能作为亲核试剂实现金催化苯酚的去芳构化反应. 2014年, Hamada小组[26]报道了第一例金催化的分子内苯酚和炔烃环化去芳构化反应, 生成有用的螺[4, 5]环己二酮结构.从苯酚对位带有炔基边链的底物33出发, 在MsOH(甲基磺酸)和2, 6-二叔丁基吡啶存在下, 在商业可得的IPrAuNTf2催化下发生5-exo-dig环化反应以50%~99%的收率得到带有环外双键螺环产物34.作者推测反应过程中首先金催化剂与底物中炔烃配位, 随后发生苯酚的原位傅克烯基化反应生成烯基金中间体15-A, 最后在MsOH作用和2, 6-二叔丁基吡啶作为缓冲剂下直接去金化生成螺环环己二烯酮34.酸的加入可以促进质子去金化过程, 碱的加入可以抑制酸催化螺环的重排反应(Scheme 15).
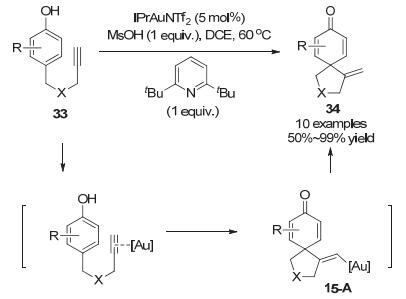 图 图式15
金催化苯酚的5-exo-dig环化反应
Figure 图式15.
Gold-catalyzed 5-exo-dig cyclization of phenols
图 图式15
金催化苯酚的5-exo-dig环化反应
Figure 图式15.
Gold-catalyzed 5-exo-dig cyclization of phenols
同一年, Vadola小组[27]报道了金催苯甲醚炔基酯35发生5-endo-dig去芳构化环化反应, 以25%~99%的收率生成螺环内酯化合物36.与之前的反应类似, 作者推测反应首先形成底物炔基与金配位的中间体16-A, 随后富电子芳环作为亲核试剂进攻被金活化的炔烃形成新的C—C键, 生成的螺环中间体16-B随后在水的存在下发生水解去甲基化, 最后中间体16-C质子化去金化生成最终产物36 (Scheme 16).
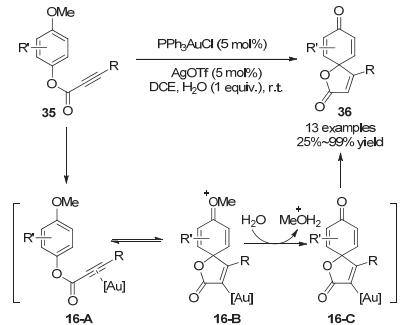 图 图式16
金催化苯甲醚的5-endo-dig环化反应
Figure 图式16.
Gold-catalyzed 5-endo-dig cyclization of aryl alkynoate esters
图 图式16
金催化苯甲醚的5-endo-dig环化反应
Figure 图式16.
Gold-catalyzed 5-endo-dig cyclization of aryl alkynoate esters
2016年, 游书力和张立明小组[28]报道了金催化萘酚去芳构化反应来直接高效地构建全碳螺环化合物38.使用商业可得的催化剂, 在简单温和的反应条件下能够快速地将1-萘酚衍生物37通过5-endo-dig环化反应转化为带有环内双键的螺环化合物38, 并且反应能够兼容卤素和各类杂环, 催化剂用量能够低至0.05 mol%, 即使敞口反应或克级规模反应都不影响反应收率.另外, 作者还尝试了反应的手性控制, 当使用手性磷酸衍生的(S)-TRIP-CPA-Ag和(4-CF3C6H4)3PAuCl作为催化剂时, 反应能够以85%的收率和90%的对映选择性得到产物38a (Scheme 17).
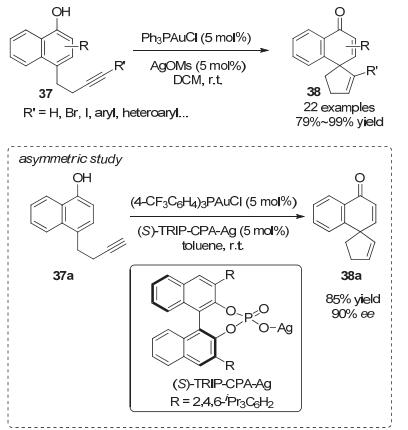 图 图式17
金催化萘酚的5-endo-dig环化反应
Figure 图式17.
Gold-catalyzed 5-endo-dig cyclization of naphthols
图 图式17
金催化萘酚的5-endo-dig环化反应
Figure 图式17.
Gold-catalyzed 5-endo-dig cyclization of naphthols
3.2 分子内环化串联去芳构化反应
在这一分类中, 大多数反应过程中也都经历了金催化的环化反应, 形成含有金的带正电荷的去芳构化中间体.为了得到稳定的产物, 在分子内引入含有亲核试剂(通常为-OH, -NHR等)的边链来捕获高活性的中间体从而生成稳定的产物, 产物一般含有多环骨架.
2010年, Wang小组[29]报道了高区域选择性(仅观察到单一异构体)的Au(Ⅰ)催化炔基吲哚串联环化的去芳构化反应, 快速高效地构建了高官能团化的多环吲哚啉类化合物.无论是炔基基团与分子内亲核试剂在同一边链的底物39, 还是炔基基团与分子内亲核试剂分别在不同边链的底物41, 在Ph3PAuSbF6的催化下都能以良好到优秀的收率(64%~88%)分别得到含有环外双键的桥环化合物40和含有环内双键的多环化合物42 (Scheme 18A).同时, 并且该方法可用于akuammiline类生物碱minfiensine的形式合成中(Scheme 18B).当使用手性富集的底物时, 在标准反应条件下能够得到对映体过量保持的产物(Scheme 18C).反应是由金催化剂活化炔烃环化启动, 吲哚作为亲核试剂进攻被配位活化的炔烃形成环化产物18-B, 分子内的亲核试剂醇羟基随后捕获亚铵成环生成氧原子桥联中间体18-C, 随后的质子化去金化生成目标产物.实验以及随后的理论计算表明[30], 吲哚N上吸电子基团对反应很重要, 能够使得生成的亚铵更为活泼, 有利于第二步分子内亲核试剂进攻成环. DFT计算表明, 与其他的烯炔环化机理不同, 反应可能是通过“两步无中间体(two-step no-intermediate)”机理进行的, 由此也说明了为何在众多可能的异构体中, 反应仅能观察到单一的异构体.
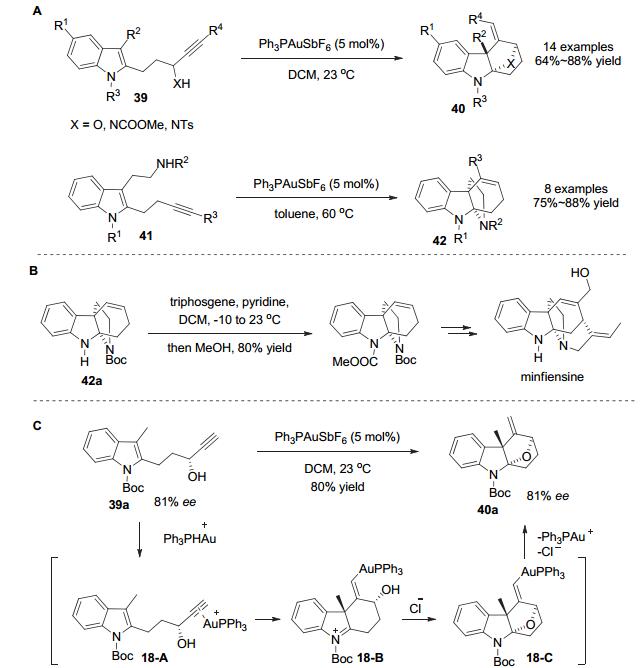 图 图式18
金催化串联环化反应构建四环吲哚啉骨架
Figure 图式18.
Gold-catalyzed tandem cyclization approach to tetracyclic indolines
图 图式18
金催化串联环化反应构建四环吲哚啉骨架
Figure 图式18.
Gold-catalyzed tandem cyclization approach to tetracyclic indolines
随后, Wang小组[31]针对炔基基团与分子内亲核试剂分别在不同边链的底物41及其金催化串联环化去芳构化反应进行了进一步的研究, 使得底物合成更快速, 反应更高效, 产物更多样.原料43可以由相应的环状亚胺与碘代炔烃烷基化得到, 经过三组分一锅反应以中等到优秀的收率(45%~83%)分别得到底物44和45 (Scheme 19A).它们分别在商业可得的金催化剂Ph3PAuNTf2催化下经历炔烃的exo-dig或endo-dig环化形成亚铵中间体, 随后被分子内的N亲核试剂捕获形成一系列并环吲哚啉化合物46和螺环吲哚啉化合物47.在这一步串联去芳构化反应中, 大多数反应中都能观察到Z1迁移到苯胺N上(Scheme 19B).接下来对并环吲哚啉化合物46和螺环吲哚啉化合物47的烷基化可以分别得到并环吲哚啉化合物48和螺环吲哚啉化合物49, 其中Z1迁移到苯胺N上的46和47先经历Z1迁回氨基N上随后苯胺N被烷基化. 46和47的还原胺化则能够得到相应的开环产物50和51 (Scheme 19C).这样, 从平面的取代吲哚经过串联金催化环化去芳构化反应以及后续的衍生化可以得到120个多样的高度官能团化的多环吲哚啉类化合物, 并用于分子药物活性测试.所得的120个多环吲哚啉化合物在MRSA(耐甲氧西林金黄色葡萄球菌)中进行筛选, 以观测其是否具有激活甲氧西林的能力.其中50a具有很好的活性, 且在进一步药效活性测试中有良好的表现, 能够激活多种β-内酰胺抗体, 为治疗MRSA感染提供可能的新选择[32].
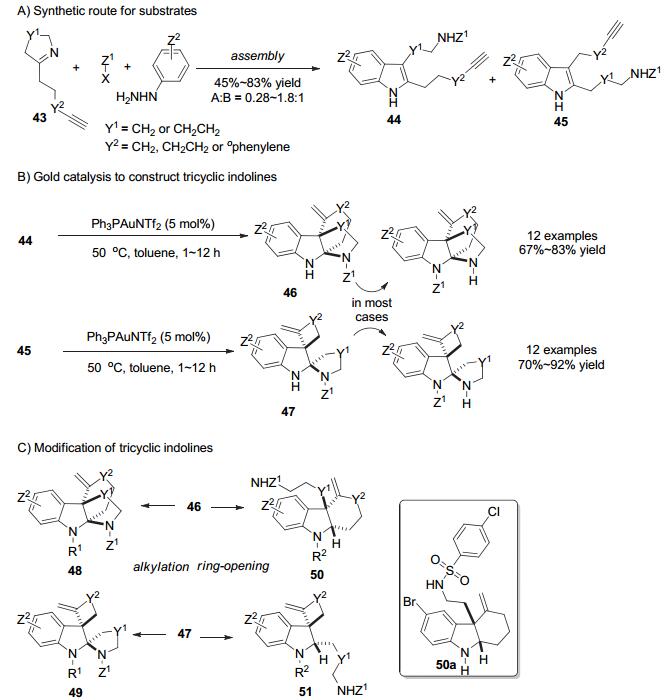 图 图式19
金催化构架多环吲哚啉类化合物的分子库
Figure 图式19.
Gold catalysis to build a library of polycyclic indolines
图 图式19
金催化构架多环吲哚啉类化合物的分子库
Figure 图式19.
Gold catalysis to build a library of polycyclic indolines
在对50a进一步的构效关系研究过程中发现50b能够给出更好的结果[33].然而由于两者较差的物理化学性质, 不适用于进一步的动物实验.在进一步优化化合物物理化学性质过程中, 作者设计在骨架中引入极性基团, 合成N杂三环吲哚啉骨架.在此目标指导下, 设计了新的底物路线, 能够得到单一异构体目标底物52.随后, 进一步拓展了金催化环化去芳构化反应的底物范围, 得到N杂三环吲哚啉类化合物53[34].与之前金催化反应条件相比, 该反应中需要使用更加富电子的膦配体XPhos (Scheme 20).
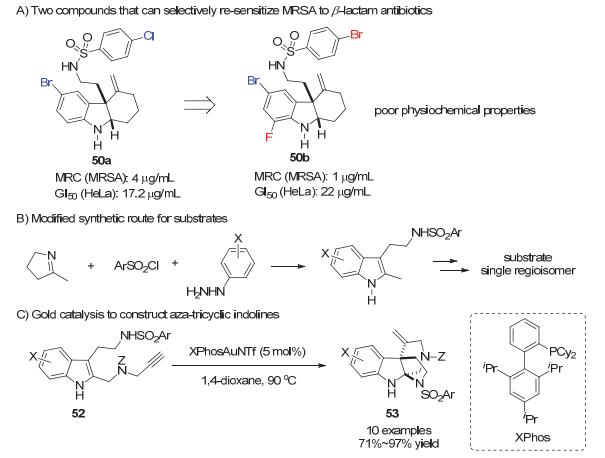 图 图式20
化合物性质导向的金催化氮杂三环吲哚啉的合成
Figure 图式20.
Property-guided synthesis of aza-tricyclic indolines: development of gold catalysis en route
图 图式20
化合物性质导向的金催化氮杂三环吲哚啉的合成
Figure 图式20.
Property-guided synthesis of aza-tricyclic indolines: development of gold catalysis en route
2011年, Bandini小组[35]也报道了金催化的吲哚类化合物去芳构化反应.相比于Wang小组合成的炔基基团与分子内亲核试剂在同一边链的底物39, Bandini小组在之前工作[36]的基础上调整了底物中分子内亲核试剂在炔基边链上的位置, 使得亲核试剂醇羟基位于边链的末端, 合成了具有不同链长吲哚类化合物54.在Echavarren金催化剂的催化下, 分别以exo-dig和endo-dig环化方式以中等到良好的收率(52%~86%)高效地分别构建了高官能团化的多环吲哚啉类化合物55和56.值得注意的是, 与Wang小组[29]报道的结果所不同的是, 吲哚N上的吸电子保护基团不是必要的.作者推测反应同样由金催化炔烃环化引发, 形成含有烯基金和亚铵的中间体.随后亚铵中间体被分子内的醇羟基捕获, 形成多环吲哚啉类化合物[37] (Scheme 21A).在之前推测的反应历程中, 第一步金催化炔烃氢-吲哚化过程是手性产生的决定步骤, 如果使用手性金催化剂, 则在理论上可以对映选择性地构建并环吲哚啉类化合物. 2012年, Bandini小组[38]实现了这样的金催化不对称去芳构化反应(Scheme 21B).在对大量反应条件筛选后, 针对5-exo-dig环化不对称去芳构化反应(R)-xylylBINAP(AuBF4)2能够给出最好的结果, 而对于7-endo-dig环化不对称去芳构化反应(S)-DTBM-Segphos(AuOTf)2能够给出最好的结果, 而两者所得产物的绝对构型是由TD-DFT计算确定的.
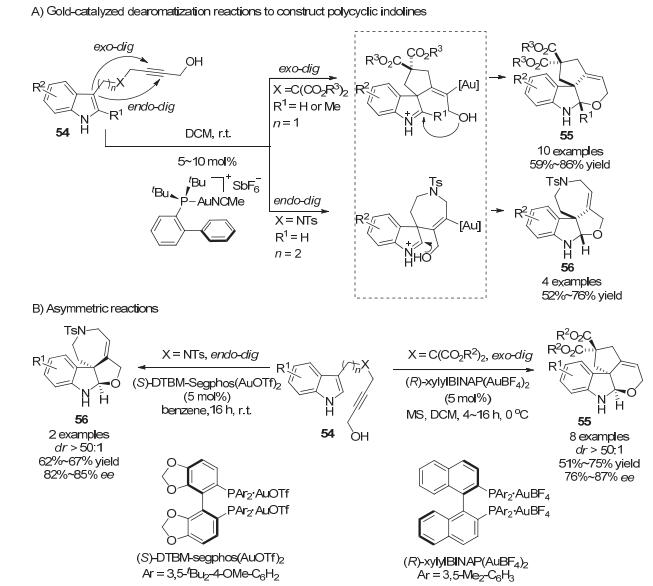 图 图式21
金催化串联反应合成四环吲哚啉类化合物及其不对称催化反应
Figure 图式21.
Synthesis of tetracyclic indolines via gold-catalyzed cascade cyclizations and enantioselective reactions
图 图式21
金催化串联反应合成四环吲哚啉类化合物及其不对称催化反应
Figure 图式21.
Synthesis of tetracyclic indolines via gold-catalyzed cascade cyclizations and enantioselective reactions
2016年, 杨震小组[39]进一步调整了吲哚底物的边链, 使用带有炔胺边链的底物57实现了金催化的环化串联去芳构化反应, 可以在温和条件下快速高效地合成四环螺吡咯吲哚啉58.对于二级醇的底物, 还可以在反应结束后低温下加入BF3•Et2O, 从而一锅生成三环螺吡咯吲哚啉59.相比于之前使用的炔丙胺边链的底物, 带有炔胺边链的底物活性更高, 反应时间更短.作者推测, 底物57中炔胺被金配位后形成烯基亚胺中间体22-A, 随后通过反式加成的形式发生环化反应形成中间体22-B, 最后分子内羟基捕获亚铵正离子中间体生成四环产物58.如果反应体系中再加入Lewis酸, 则会促进hemi-aminol开环生成有Lewis酸配位的中间体22-C, 随后发生非对映选择性的[1, 5]-H迁移反应生成三环产物59.另外, 作者还尝试了反应的不对称控制, 当使用商业可得的手性双膦联苯配体(R)-L2时, 能够以85%收率和60% ee对映选择性得到产物58a (Scheme 22).
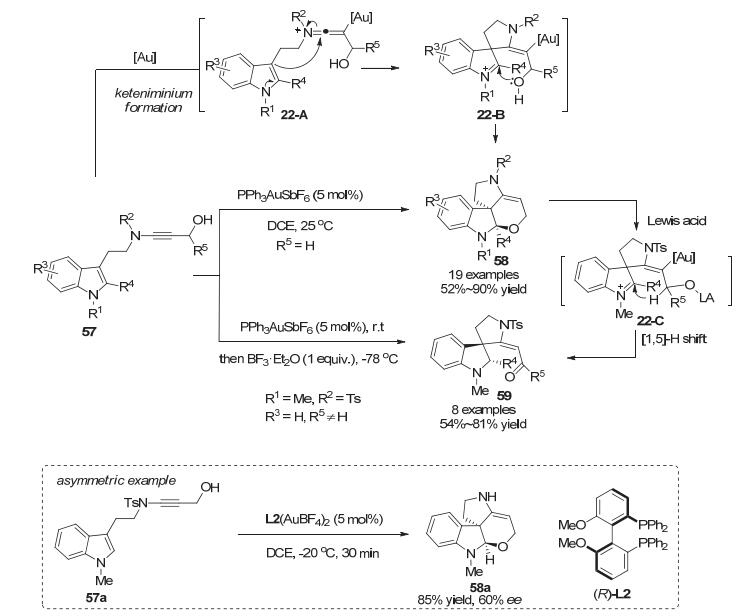 图 图式22
金催化吲哚-炔胺底物的分子内串联环化反应
Figure 图式22.
Gold-catalyzed intramolecular tandem cyclization of indole-ynamides
图 图式22
金催化吲哚-炔胺底物的分子内串联环化反应
Figure 图式22.
Gold-catalyzed intramolecular tandem cyclization of indole-ynamides
2012年, Van der Eycken小组[40]报道了Ugi金催化的多米诺环化反应, 对四组分反应生成的取代吲哚60进行去芳构化反应, 快速高效地构建了多并环螺-吲哚啉类化合物61.第一步的Ugi四组分反应从简单易得的原料出发, 以中等到良好的收率(45%~87%)得到吲哚C3官能团化的底物60[41], 为后续金催化去芳构化反应提供了反应启动位点“炔基”和后续捕获中间体的分子内亲核试剂“N—H”.这与之前的文献报道的策略不同, 采用此处的“branched-handed”策略, 即C3边链分支成两条边链, 分别含有炔基和分子内亲核试剂(Scheme 23A). 2013年, Van der Eycken小组[42]进一步拓展该反应的底物, 在Ugi四组分反应中使用不同的组合, 得到不同官能团化的底物.在之前的反应中使用丙炔酸衍生物和烷基胺, 在此处使用羧酸和炔丙胺, 合成结构更丰富的底物.同样在金催化下发生去芳构化反应以中等到优秀的收率(40%~84%)得到并环吲哚啉类产物62.其中, TFA的加入能够提高收率(Scheme 23B).
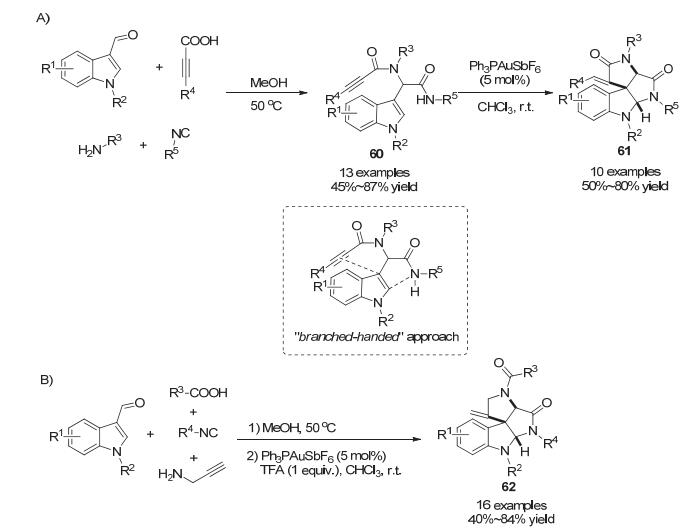 图 图式23
Ugi/Au(Ⅰ)催化的多米诺环化反应合成螺-吲哚啉类化合物
Figure 图式23.
Ugi/gold-catalyzed diastereoselective domino cyclization
图 图式23
Ugi/Au(Ⅰ)催化的多米诺环化反应合成螺-吲哚啉类化合物
Figure 图式23.
Ugi/gold-catalyzed diastereoselective domino cyclization
众所周知, 金催化苯胺的邻位炔基环化反应能够高效构建吲哚环.如果一个金络合物能够同时催化炔烃环化生成吲哚环以及生成的吲哚进攻另一个被金活化的炔烃/联烯发生环化去芳构化反应, 则可以从简单的邻位炔基苯胺类底物快速、高效、直接地构建具有三维立体结构的并环骨架. 2015年, Fujii和Ohno小组[43]从2-烷基炔-N-炔丙基苯胺类底物63出发实现了这样的串联反应, 以中等到优秀的收率(59%~94%)得到四环并吲哚啉产物64.作者推测在串联反应中存在两个催化循环.在金催化吲哚环构建过程中(Cycle A), 苯胺N原子亲核进攻被金活化的炔烃从而发生环化反应形成吲哚-金中间体24-B, 随后N上的炔丙基经[1, 3]-迁移到吲哚C3位生成联烯65[44], 并且这样的联烯中间体可以被分离鉴定.体系中生成的联烯会被金催化剂活化发生环化反应得到中间体24-D, 随后扩环生成烯基金中间体24-E, 这样的中间体可以被烯基金所稳定形成24-E', 最后经历分子内亲核加成和质子化去金化得到并环吲哚啉产物64. NMR实验表明, 即使形成吲哚环(24-A到24-B)是一个较快的过程, 但由于炔丙基重排反应(24-B到65)是一个较慢的过程, 因此催化循环Ⅰ要比催化循环Ⅱ慢得多.作者推测炔丙基重排反应(24-B到65)可能是反应的决速步, 并且进一步实验结果表明这是一个分子内的重排反应(Scheme 24).
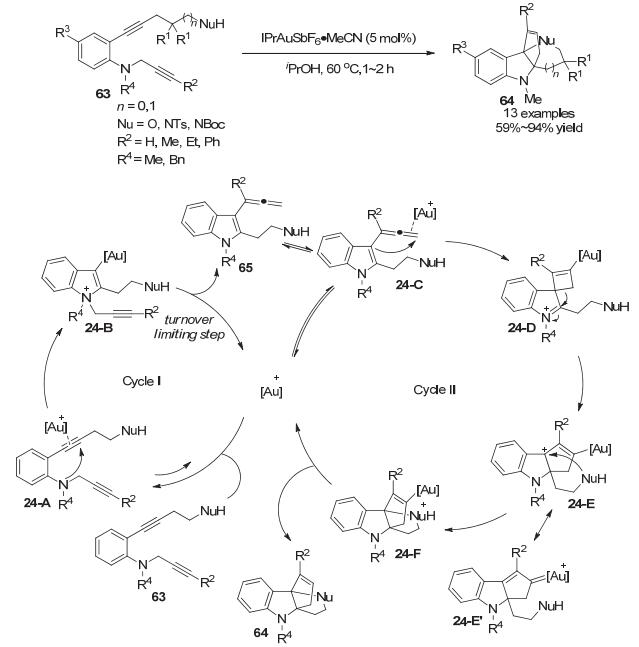 图 图式24
经炔丙基重排的金催化2-炔基-N-炔丙基苯胺的串联环化反应
Figure 图式24.
Gold-catalyzed cascade cyclization of 2-alkynyl-N-propargylanilines by rearrangement of a propargyl group
图 图式24
经炔丙基重排的金催化2-炔基-N-炔丙基苯胺的串联环化反应
Figure 图式24.
Gold-catalyzed cascade cyclization of 2-alkynyl-N-propargylanilines by rearrangement of a propargyl group
除了只用含有N/O原子的亲核试剂来捕获金催化炔烃/联烯环化后生成中间体以外, 还可以使用含有C亲核试剂来实现金催化环化串联去芳构化反应.在Tanaka小组[45]前期的工作中, 通过Pt催化的炔烃环化反应实现了萘环去芳构化(Scheme 25, Eq. 1).作者推测在反应过程中, 原位生成的阳离子Pt催化剂与炔烃配位发生endo环化生成中间体25-A, 随后骨架重排经中间体25-B生成中间体25-C, 但也不排除中间体25-C是从底物66直接原位环化得到的.随后中间体25-C经傅克类型反应、去质子以及质子化去金属化得到多环产物67. 2015年, Tanaka小组[46]改变了底物66中炔基和发生傅克反应的富电子芳环的位置合成了底物68, 利用手性金络合物实现了不对称催化反应, 能够以中等到良好的收率和对映选择性得到产物69和69' (产物比例与取代基R'有关)(Scheme 25, Eq. 2).在进一步的底物拓展中, 作者将富电子芳环换为芳杂环以及改变边链上官能团连接顺序, 反应也能够顺利发生[47] (Scheme 25, Eq. 3 & 4).
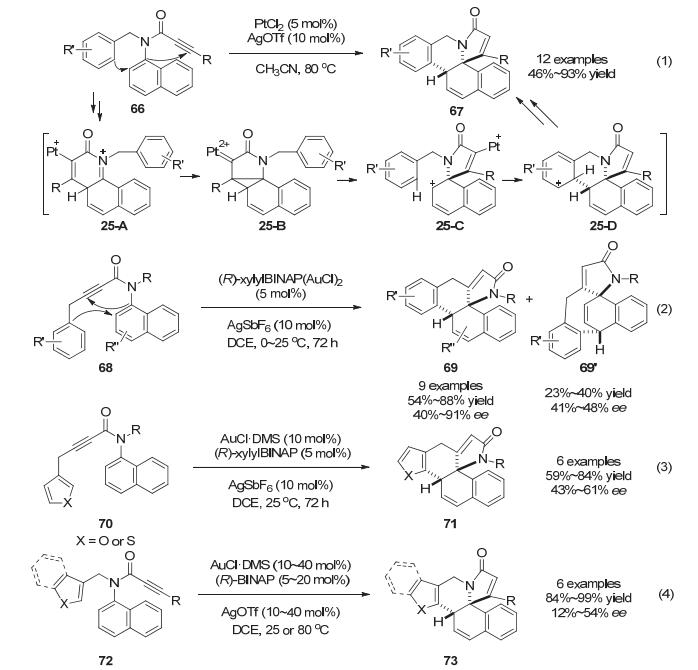 图 图式25
金催化1-萘胺的环化反应
Figure 图式25.
Gold-catalyzed cyclization of 1-aminonaphthalenes
图 图式25
金催化1-萘胺的环化反应
Figure 图式25.
Gold-catalyzed cyclization of 1-aminonaphthalenes
3.3 分子间去芳构化反应
之前的金催化氢-官能团化反应都是将含有炔烃边链引入底物, 从而实现金催化的分子内去芳构化反应.除此之外, 还可以调节炔烃/联烯和芳环亲核试剂的活性, 实现金催化分子间的去芳构化反应.分子间的反应难度在于反应的化学选择性和区域选择性.另外, 去芳构化过程本身是相对困难的, 需要克服一定的芳香性能垒, 一般反应条件相对剧烈.在剧烈的条件下, 要调控反应化学选择性和区域选择性, 无疑是十分具有挑战性的.
2014年, Bandini小组[48]使用2, 3-二取代吲哚74作为底物, 使之与保护的联烯酰胺75发生分子间反应, 在双官能团催化剂(2, 4-tBu2C6H3O)3PAuTFA存在下能够以良好到优秀的收率得到亚胺产物76.作者认为在反应过程中, 双官能团催化剂一方面作为Lewis酸活化联烯的π体系, 另一方面阴离子TFA-作为Brϕnsted碱通过氢键活化吲哚, 使得反应有专一的区域选择性(Scheme 26).
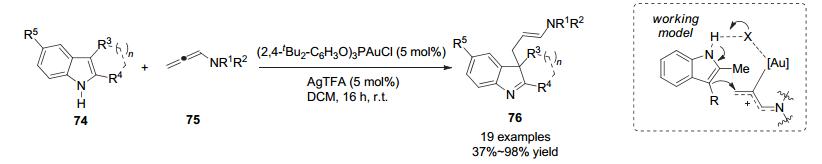 图 图式26
金催化吲哚与联烯酰胺分子间去芳构化反应
Figure 图式26.
Taming gold(Ⅰ)-counterion interplay in the dearomatization of indoles with allenamides
图 图式26
金催化吲哚与联烯酰胺分子间去芳构化反应
Figure 图式26.
Taming gold(Ⅰ)-counterion interplay in the dearomatization of indoles with allenamides
吲哚与联烯酰胺发生分子间去芳构化后生成的亚铵中间体可以被分子内的亲核试剂捕获, 形成并环化合物. 2015年, 陈自立小组[49]使用色胺或色醇77作为底物, 在手性金络合物催化剂存在下, 与联烯酰胺75反应, 以中等到优秀的对映选择性快速构建吡咯吲哚啉类化合物78 (Scheme 27).
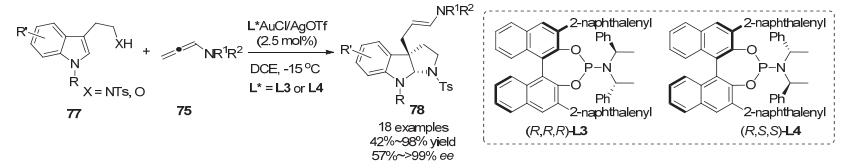 图 图式27
不对称金催化吲哚与联烯酰胺分子间串联去芳构化反应
Figure 图式27.
Enantioselective gold-catalyzed cascade construction of pyrroloindoline derivatives
图 图式27
不对称金催化吲哚与联烯酰胺分子间串联去芳构化反应
Figure 图式27.
Enantioselective gold-catalyzed cascade construction of pyrroloindoline derivatives
在之前的金催化吲哚分子间去芳构化生成亚铵中间体28-A经历质子去金化而生成最终产物[48].基于此, 作者设想是否能够通过调节反应底物结构和金催化剂的电性来抑制中间体28-A质子去金化过程, 从而使中间体28-A有更多的转化途径. 2015年, Bandini小组[50]使用吲哚N上带有吸电子取代基的底物79和取代联烯酰胺75a, 并且使用富电子的手性配体(R)-DTBM-Segphos, 实现了第二步关环过程, 以优秀的对映选择性(81%~99% ee)生成带有2个连续手性季碳中心和并环四元环吲哚啉结构的[2+2]-环加成动力学稳定产物80.吲哚N上带有的吸电子取代基可以提高假吲哚中间体28-A的亲电性, 富电子的配体可以增加烯基金物种的亲核性, 从而协助了第二步关环过程.进一步研究表明[51], 当使用活性较低的联烯基酯81时, 需要适当提高温度, 也可以中等到优秀的对映选择性得到[2+2]-环加成产物82.理论计算阐释了反应中去芳构化形成中间体28-A是决速步, 第二步关环涉及烯基C—Au键的断裂过程, 由此保持了双键的Z构型(Scheme 28).
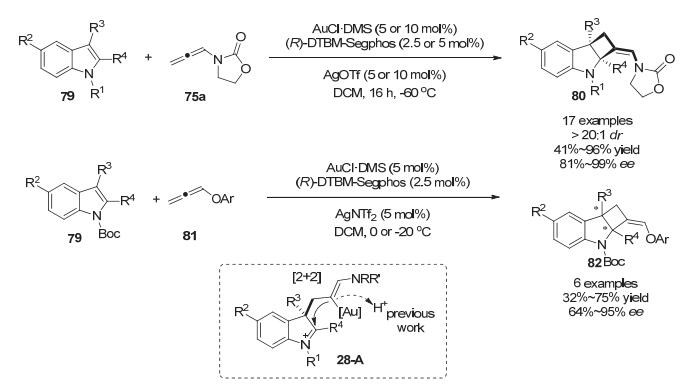 图 图式28
不对称金催化吲哚与联烯酰胺[2+2]环加成反应
Figure 图式28.
Enantioselective gold-catalyzed dearomative [2+2]-cycloaddition between indoles and allenamides
图 图式28
不对称金催化吲哚与联烯酰胺[2+2]环加成反应
Figure 图式28.
Enantioselective gold-catalyzed dearomative [2+2]-cycloaddition between indoles and allenamides
2013年, Vicente小组[52]使用2-烯基取代的吲哚底物83与联烯酰胺75反应, 在温和条件下完成了金催化的分子间[4+2]-环加成反应, 实现了吲哚分子间去芳构化反应, 以中等到优秀的收率得到了四氢咔唑骨架的产物84.如果使用3-烯基取代的吲哚底物85在AuCl3催化下反应也能顺利进行, 以93%的收率得到环加成产物86 (Scheme 29).
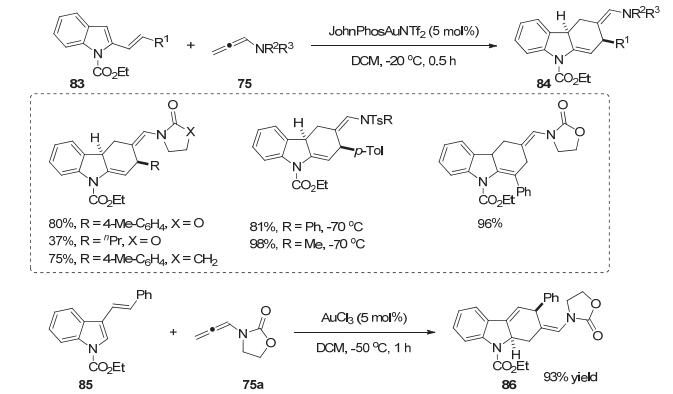 图 图式29
金催化烯基吲哚与联烯酰胺分子间[4+2]环加成反应
Figure 图式29.
Gold-catalyzed intermolecular [4+2]-cycloaddition of vinyl indoles and N-allenamides
图 图式29
金催化烯基吲哚与联烯酰胺分子间[4+2]环加成反应
Figure 图式29.
Gold-catalyzed intermolecular [4+2]-cycloaddition of vinyl indoles and N-allenamides
2015年, 张俊良小组[53]使用手性亚磷酰胺配体实现了不对称的金催化[4+2]-环加成去芳构化反应.当3-烯基取代吲哚底物87吲哚N上的取代基为吸电子取代基时, 在2.5 mol%手性金催化剂存在下, 能够以优秀的对映选择性和收率快速转化为多环产物89, Z/E选择性最高可达14 : 1 (Scheme 30).
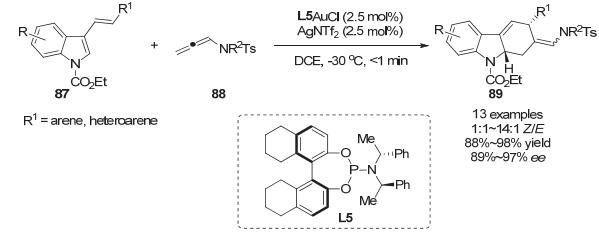 图 图式30
不对称金催化苯乙烯基吲哚与联烯酰胺分子间[4+2]环加成反应
Figure 图式30.
Enantioselective gold-catalyzed intermolecular [4+2]-cycloadditions of 3-styrylindoles with N-allenamides
图 图式30
不对称金催化苯乙烯基吲哚与联烯酰胺分子间[4+2]环加成反应
Figure 图式30.
Enantioselective gold-catalyzed intermolecular [4+2]-cycloadditions of 3-styrylindoles with N-allenamides
4 总结与展望
虽然金催化去芳构化反应起步较晚, 但已经展现出了非常大的潜力.并且反应条件一般比较温和, 反应快速高效, 对于官能团具有较好的兼容性, 并且也有一些不对称催化的例子给出不错的对映选择性.目前, 金催化去芳构化反应的反应模式还比较单一, 主要局限于金催化的重排反应和炔烃/联烯的氢-官能团化反应.其中, 通过重排反应实现的金催化芳构化反应的例子相对较为零散, 仍有非常大的发展潜力, 而通过炔烃的氢-官能团化反应实现的金催化芳构化反应的例子虽然较多, 但使用的底物还相对简单, 目前使用较多的底物集中于吲哚、苯酚和萘酚.另外, 金催化反应的手性控制也相对困难, 目前对映选择性较好的结果还比较少.
可以预见在不远的将来, 随着更多金催化模式被发现和发展, 以及手性控制策略的发展, 将会有更多的金催化反应来实现去芳构化过程, 并且反应的手性控制也会有更多的方法, 也会有更多的金催化去芳构化反应被应用于复杂的天然产物和生物活性分子的合成中.
-
-
[1]
Ito, Y.; Sawamura, M.; Hayashi, T. J. Am. Chem. Soc. 1986, 108, 6405. doi: 10.1021/ja00280a056
-
[2]
(a) Teles, J. H.; Brode, S.; Chabanas, M. Angew. Chem., Int. Ed. 1998, 37, 1415; (b) Mizushima, E.; Sato, K.; Hayashi, T.; Tanaka, M. Angew. Chem., Int. Ed. 2002, 41, 4563.
-
[3]
For recent books: (a) Toste, F. D.; Michelet, V. Gold Catalysis: An Homogeneous Approach, Imperial College Press, London, 2014; (b) Slaughter, L. M. Homogeneous Gold Catalysis, Springer, 2015; (c) Rappoport, Z.; Liebman, J. F.; Marek, I. The Chemistry of Organogold Compounds, Wiley, Chichester, 2014.
-
[4]
For recent reviews: (a) Hashmi, A. S. K. Acc. Chem. Res. 2014, 47, 864; (b) Yeom, H.-S.; Shin, S. Acc. Chem. Res. 2014, 47, 966; (c) Zhang, L. Acc. Chem. Res. 2014, 47, 877; (d) Wang, Y.-M.; Lackner, A. D.; Toste, F. D. Acc. Chem. Res. 2014, 47, 889; (e) Obradors, C.; Echavarren, A. M. Acc. Chem. Res. 2014, 47, 902; (f) Zhang, D.-H.; Tang, X.-Y.; Shi, M. Acc. Chem. Res. 2014, 47, 913; (g) Yang, W.; Hashmi, A. S. K. Chem. Soc. Rev. 2014, 43, 2941; (h) Xie, J.; Pan, C.; Abdukader, A.; Zhu, C. Chem. Soc. Rev. 2014, 43, 5245; (i) Muratore, M. E.; Homs, A.; Obradors, C.; Echavarren, A. M. Chem. Asian J. 2014, 9, 3066; (j) Inamdar, S. M.; Konala, A.; Patil, N. T. Chem. Commun. 2014, 50, 15124; (k) Obradors, C.; Echavarren, A. M. Chem. Commun. 2014, 50, 16; (l) Gu, P.; Xu, Q.; Shi, M. Tetrahedron Lett. 2014, 55, 577; (m) Qian, D.; Zhang, J. Chem. Soc. Rev. 2015, 44, 677; (n) Joost, M.; Amgoune, A.; Bourissou, D. Angew. Chem., Int. Ed. 2015, 54, 15022; (o) Jia, M.; Bandini, M. ACS Catal. 2015, 5, 1638; (p) Debrouwer, W.; Heugebaert, T. S. A.; Roman, B. I.; Stevens, C. V. Adv. Synth. Catal. 2015, 357, 2975; (q) Goodwin, J. A.; Aponick, A. Chem. Commun. 2015, 51, 8730; (r) Dorel, R.; Echavarren, A. M. J. Org. Chem. 2015, 80, 7321; (s) Ranieri, B.; Escofet, I.; Echavarren, A. M. Org. Biomol. Chem. 2015, 13, 7103; (t) Wei, F.; Song, C.; Ma, Y.; Zhou, L.; Tung, C.-H.; Xu, Z. Sci. Bull. 2015, 60, 1479; (u) Liu, L.; Zhang, J. Chem. Soc. Rev. 2016, 45, 506; (v) Zheng, Z.; Wang, Z.; Wang, Y.; Zhang, L. Chem. Soc. Rev. 2016, 45, 4448; (w) Zi, W.; Dean, T. F. Chem. Soc. Rev. 2016, 45, 4567; (x) Li, Y.; Li, W.; Zhang, J. Chem. Eur. J. 2017, 23, 467.
-
[5]
(a) Hashmi, A. S. K.; Rudolph, M. Chem. Soc. Rev. 2008, 37, 1766; (b) Alcaide, B.; Almendros, P.; Alonso, J. M. Molecules 2011, 16, 7815; (c) Rudolph, M.; Hashmi, A. S. K. Chem. Soc. Rev. 2012, 41, 2448; (d) Barbour, P. M.; Marholz, L. J.; Chang, L.; Xu, W.; Wang, X. Chem. Lett. 2014, 43, 572; (e) Fensterbank, L.; Malacria, M. Acc. Chem. Res. 2014, 47, 953; (f) Füerstner, A. Angew. Chem., Int. Ed. 2014, 53, 8587; (g) Füerstner, A. Acc. Chem. Res. 2014, 47, 925; (h) Zhang, Y.; Luo, T.; Yang, Z. Nat. Prod. Rep. 2014, 31, 489; (i) Pflästerer, D.; Hashmi, A. S. K. Chem. Soc. Rev. 2016, 45, 1331.
-
[6]
Gorin, D. J.; Toste, F. D. Nature 2007, 446, 395. doi: 10.1038/nature05592
-
[7]
For a recent book: (a) Hashmi, A. S. K. ; Toste, D. F. ; Toste, F. D. Modern Gold Catalyzed Synthesis, Wiley, 2012; For recent selected examples: (b) Chen, B. ; Yu, C. ; Zhang, G. Chin. J. Org. Chem. 2015, 35, 625 (陈斌, 于丛军, 张国柱, 有机化学, 2015, 35, 625); (c) Li, L. ; Zhou, B. ; Ye, L. -W. Chin. J. Org. Chem. 2015, 35, 655 (李龙, 周波, 叶龙武, 有机化学, 2015, 35, 655); (d) Zhang, X. ; Sun, X. ; Cui, X. ; Zhang, H. Chin. J. Org. Chem. 2015, 35, 1700 (张小祥, 孙小萍, 崔杏丽, 张海飞, 有机化学, 2015, 35, 1700); (e) Zhang, X. ; Sun, X. ; Zhang, H. ; Cui, X. ; Ma, M. Chin. J. Org. Chem. 2015, 35, 1469 (张小祥, 孙小萍, 张海飞, 崔杏丽, 马猛涛, 有机化学, 2015, 35, 1469); (f) Li, X. -L. ; Wang, J. -Q. ; Li, L. ; Yin, Y. -W. ; Ye, L. -W. Acta Chim. Sinica. 2016, 74, 49 (李新玲, 王佳琪, 李龙, 尹应武, 叶龙武, 化学学报, 2016, 74, 49).
-
[8]
For recent reviews: (a) Pape, A. R. ; Kaliappan, K. P. ; Kündig, E. P. Chem. Rev. 2000, 100, 2917; (b) Zhuo, C. -X. ; Zhang, W. ; You, S. -L. Angew. Chem. , Int. Ed. 2012, 51, 12662; (c) Zhuo, C. -X. ; Zheng, C. ; You, S. -L. Acc. Chem. Res. 2014, 47, 2558; (d) Wu, W. -T. ; Zhang, L. ; You, S. -L. Chem. Soc. Rev. 2016, 45, 1570; (e) Sun, W. ; Li, G. ; Hong, L. ; Wang, R. Org. Biomol. Chem. 2016, 14, 2164; (f) Zheng, C. ; You, S. -L. Chem. 2016, 1, 830; For recent selected examples: (g) Duan, D. -H. ; Yin, Q. ; Wang, S. -G; Gu, Q. ; You, S. -L. Acta Chim. Sinica. 2014, 72, 1001 (段德河, 殷勤, 王守国, 顾庆, 游书力, 化学学报, 2014, 72, 1001); (h) Wang, Y. ; Liu, R. ; Gao, J. ; Jia, Y. Chin. J. Org. Chem. 2017, 37, 691 (王永刚, 刘人荣, 高建荣, 贾义霞, 有机化学, 2017, 37, 691).
-
[9]
(a) Krause, N.; Winter, C. Chem. Rev. 2011, 111, 1994; (b) Rudolph, M.; Hashmi, A. S. K. Chem. Commun. 2011, 47, 6536; (c) Gulevich, A. V.; Dudnik, A. S.; Chernyak, N.; Gevorgyan, V. Chem. Rev. 2013, 113, 3084; (d) Qian, D.; Zhang, J. Chem. Rec. 2014, 14, 280; (e) Wei, Y.; Shi, M. ACS Catal. 2016, 2515.
-
[10]
Bandini, M. Chem. Soc. Rev. 2011, 40, 1358. doi: 10.1039/C0CS00041H
-
[11]
Zhang, L. J. Am. Chem. Soc. 2005, 127, 16804. doi: 10.1021/ja056419c
-
[12]
Yang, J.-M.; Li, P.-H.; Wei, Y.; Tang, X.-Y.; Shi, M. Chem. Commun. 2016, 52, 346. doi: 10.1039/C5CC08381H
-
[13]
Zi, W.; Wu, H.; Toste, F. D. J. Am. Chem. Soc. 2015, 137, 3225. doi: 10.1021/jacs.5b00613
-
[14]
Zhang, G.; Huang, X.; Li, G.; Zhang, L. J. Am. Chem. Soc. 2008, 130, 1814. doi: 10.1021/ja077948e
-
[15]
Zhang, G.; Zhang, L. J. Am. Chem. Soc. 2008, 130, 12598. doi: 10.1021/ja804690u
-
[16]
Briones, J. F.; Davies, H. M. L. J. Am. Chem. Soc. 2012, 134, 11916. doi: 10.1021/ja304506g
-
[17]
Tokimizu, Y.; Oishi, S.; Fujii, N.; Ohno, H. Org. Lett. 2014, 16, 3138. doi: 10.1021/ol5012604
-
[18]
(a) Nevado, C.; Echavarren, A. M. Synthesis 2005, 167; (b) Kitamura, T. Eur. J. Org. Chem. 2009, 2009, 1111; (d) Yamamoto, Y. Chem. Soc. Rev. 2014, 43, 1575.
-
[19]
Ferrer, C.; Echavarren, A. M. Angew. Chem., Int. Ed. 2006, 45, 1105. doi: 10.1002/(ISSN)1521-3773
-
[20]
Ferrer, C.; Amijs, C. H. M.; Echavarren, A. M. Chem. Eur. J. 2007, 13, 1358. doi: 10.1002/(ISSN)1521-3765
-
[21]
Zhang, Y.-Q.; Zhu, D.-Y.; Jiao, Z.-W.; Li, B.-S.; Zhang, F.-M.; Tu, Y.-Q.; Bi, Z. Org. Lett. 2011, 13, 3458. doi: 10.1021/ol201194n
-
[22]
Cheng, B.; Huang, G.; Xu, L.; Xia, Y. Org. Biomol. Chem. 2012, 10, 4417. doi: 10.1039/c2ob25316j
-
[23]
Xu, W.; Wang, W.; Wang, X. Angew. Chem., Int. Ed. 2015, 54, 9546. doi: 10.1002/anie.v54.33
-
[24]
Nishiyama, D.; Ohara, A.; Chiba, H.; Kumagai, H.; Oishi, S.; Fujii, N.; Ohno, H. Org. Lett. 2016, 18, 1670. doi: 10.1021/acs.orglett.6b00536
-
[25]
Zhang, L.; Wang, Y.; Yao, Z. J.; Wang, S.; Yu, Z.-X. J. Am. Chem. Soc. 2015, 137, 13290. doi: 10.1021/jacs.5b05971
-
[26]
Nemoto, T.; Matsuo, N.; Hamada, Y. Adv. Synth. Catal. 2014, 356, 2417. doi: 10.1002/adsc.v356.11/12
-
[27]
Aparece, M. D.; Vadola, P. A. Org. Lett. 2014, 16, 6008. doi: 10.1021/ol503022h
-
[28]
Wu, W.-T.; Xu, R.-Q.; Zhang, L.; You, S.-L. Chem. Sci. 2016, 7, 3427. doi: 10.1039/C5SC04130A
-
[29]
Liu, Y.; Xu, W.; Wang, X. Org. Lett. 2010, 12, 1448. doi: 10.1021/ol100153h
-
[30]
Noey, E. L.; Wang, X.; Houk, K. N. J. Org. Chem. 2011, 76, 3477. doi: 10.1021/jo200556f
-
[31]
Podoll, J. D.; Liu, Y.; Chang, L.; Walls, S.; Wang, W.; Wang, X. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 15573. doi: 10.1073/pnas.1310459110
-
[32]
Barbour, P. M.; Podoll, J. D.; Marholz, L. J.; Wang, X. Bioorg. Med. Chem. Lett. 2014, 24, 5602. doi: 10.1016/j.bmcl.2014.10.094
-
[33]
Chang, L.; Podoll, J. D.; Wang, W.; Walls, S.; O'Rourke, C. P.; Wang, X. J. Med. Chem. 2014, 57, 3803. doi: 10.1021/jm500146g
-
[34]
Barbour, P. M.; Wang, W.; Chang, L.; Pickard, K. L.; Rais, R.; Slusher, B. S.; Wang, X. Adv. Synth. Catal. 2016, 358, 1482. doi: 10.1002/adsc.v358.9
-
[35]
Cera, G.; Crispino, P.; Monari, M.; Bandini, M. Chem. Commun. 2011, 47, 7803. doi: 10.1039/c1cc12328a
-
[36]
Bandini, M.; Eichholzer, A. Angew. Chem., Int. Ed. 2009, 48, 9533. doi: 10.1002/anie.200904388
-
[37]
Cera, G.; Chiarucci, M.; Bandini, M. Pure Appl. Chem. 2012, 84, 1673.
-
[38]
Cera, G.; Chiarucci, M.; Mazzanti, A.; Mancinelli, M.; Bandini, M. Org. Lett. 2012, 14, 1350. doi: 10.1021/ol300297t
-
[39]
Zheng, N.; Chang, Y.-Y.; Zhang, L.-J.; Gong, J.-X.; Yang, Z. Chem. Asian J. 2016, 11, 371. doi: 10.1002/asia.v11.3
-
[40]
Modha, S. G.; Kumar, A.; Vachhani, D. D.; Jacobs, J.; Sharma, S. K.; Parmar, V. S.; Van Meervelt, L.; Van der Eycken, E. V. Angew. Chem., Int. Ed. 2012, 51, 9572. doi: 10.1002/anie.v51.38
-
[41]
Modha, S. G.; Vachhani, D. D.; Jacobs, J.; Van Meervelt, L.; Van der Eycken, E. V. Chem. Commun. 2012, 48, 6550. doi: 10.1039/c2cc32586a
-
[42]
Kumar, A.; Vachhani, D. D.; Modha, S. G.; Sharma, S. K.; Parmar, V. S.; Van der Eycken, E. V. Beilstein J. Org. Chem. 2013, 9, 2097. doi: 10.3762/bjoc.9.246
-
[43]
Tokimizu, Y.; Oishi, S.; Fujii, N.; Ohno, H. Angew. Chem., Int. Ed. 2015, 54, 7862. doi: 10.1002/anie.201502256
-
[44]
审稿人认为N上炔丙基的迁移在形式上更倾向于[3, 3]-重排的过程.
-
[45]
Shibuya, T.; Noguchi, K.; Tanaka, K. Angew. Chem., Int. Ed. 2012, 51, 6219. doi: 10.1002/anie.201202165
-
[46]
Oka, J.; Okamoto, R.; Noguchi, K.; Tanaka, K. Org. Lett. 2015, 17, 676. doi: 10.1021/ol503698s
-
[47]
Baba, T.; Oka, J.; Noguchi, K.; Tanaka, K. Eur. J. Org. Chem. 2015, 2015, 4374. doi: 10.1002/ejoc.201500486
-
[48]
Jia, M.; Cera, G.; Perrotta, D.; Monari, M.; Bandini, M. Chem. Eur. J. 2014, 20, 9875. doi: 10.1002/chem.201403155
-
[49]
Shen, Z.-Q.; Li, X.-X.; Shi, J.-W.; Chen, B.-L.; Chen, Z. Tetrahedron Lett. 2015, 56, 4080. doi: 10.1016/j.tetlet.2015.05.021
-
[50]
Jia, M.; Monari, M.; Yang, Q.-Q.; Bandini, M. Chem. Commun. 2015, 51, 2320. doi: 10.1039/C4CC08736D
-
[51]
Ocello, R.; De Nisi, A.; Jia, M.; Yang, Q.-Q.; Monari, M.; Giacinto, P.; Bottoni, A.; Miscione, G. P.; Bandini, M. Chem. Eur. J. 2015, 21, 18445. doi: 10.1002/chem.201503598
-
[52]
Pirovano, V.; Decataldo, L.; Rossi, E.; Vicente, R. Chem. Commun. 2013, 49, 3594. doi: 10.1039/c3cc41514g
-
[53]
Wang, Y.; Zhang, P.; Liu, Y.; Xia, F.; Zhang, J. Chem. Sci. 2015, 6, 5564. doi: 10.1039/C5SC01827G
-
[1]
-

图式1 金催化的炔丙酯及其衍生物的重排反应的两条可能反应路径
Scheme 1 Two pathways for gold-catalyzed rearrangements of propargyl ester and its derivatives

图式2 金催化炔丙酯[3, 3]-迁移重排[2+2]环加成串联反应
Scheme 2 Tandem Au-catalyzed 1, 3-rearrangement-[2+2] cycloadditions of propargylic esters

图式3 金催化炔丙酯[1, 2]-迁移重排[3+2]环加成串联反应
Scheme 3 Tandem Au-catalyzed 1, 2-rearrangement-[3+2] cycloadditions of propargylic esters

图式4 不对称金催化去芳构化Rautenstrauch重排反应
Scheme 4 Gold(Ⅰ)-catalyzed asymmetric dearomative Rautenstrauch rearrangement

图式5 金催化重排反应生成的含金全碳1, 4-偶极体参与的[4+2]环化反应
Scheme 5 Au-Containing all-carbon 1, 4-dipoles: generation and [4+2] annulation

图式6 金催化重排反应生成的含金全碳1, 3-偶极体参与的[3+2]环化反应
Scheme 6 Au-containing all-carbon 1, 3-dipoles: generation and [3+2] cycloaddition reactions

图式7 金催化内炔烃不对称环丙烯化反应研究中意外发现的重排反应
Scheme 7 Unexpected rearrangement in the study of gold(Ⅰ)-catalyzed asymmetric cyclopropenation of internal alkynes

图式9 金催化炔烃的氢-官能团化反应模式及其在吲哚合成和修饰中的应用
Scheme 9 General model for gold-catalyzed hydrofunctionalization of alkynes and its application in the synthesis and decoration of indoles

图式10 第一例通过金催化炔烃的氢-官能团化反应实现的去芳构化反应
Scheme 10 First example of dearomatization reaction via gold-catalyzed hydrofunctionalization of alkynes

图式11 金催化吲哚C2位环化C3位苯酚氧基离去反应
Scheme 11 Gold-catalyzed indole C2-site annulation and removal of phenoxy group at the C3-site

图式12 首例使用吲哚N硅基保护衍生物的金催化去芳构化环化反应
Scheme 12 First report of gold-catalyzed dearomative cyclization of N-silyl indole derivatives

图式13 基于金催化环化反应的(±)-strictamine的形式全合成
Scheme 13 Formal total synthesis of (±)-strictamine based on a gold-catalyzed cyclization

图式14 金催化2-炔丙基-β-四氢咔唑的扩环和螺环化反应
Scheme 14 Gold-catalyzed ring expansion and spirocyclization of 2-propargyl-β-tetrahydrocarbolines

图式16 金催化苯甲醚的5-endo-dig环化反应
Scheme 16 Gold-catalyzed 5-endo-dig cyclization of aryl alkynoate esters

图式17 金催化萘酚的5-endo-dig环化反应
Scheme 17 Gold-catalyzed 5-endo-dig cyclization of naphthols

图式18 金催化串联环化反应构建四环吲哚啉骨架
Scheme 18 Gold-catalyzed tandem cyclization approach to tetracyclic indolines

图式19 金催化构架多环吲哚啉类化合物的分子库
Scheme 19 Gold catalysis to build a library of polycyclic indolines

图式20 化合物性质导向的金催化氮杂三环吲哚啉的合成
Scheme 20 Property-guided synthesis of aza-tricyclic indolines: development of gold catalysis en route

图式21 金催化串联反应合成四环吲哚啉类化合物及其不对称催化反应
Scheme 21 Synthesis of tetracyclic indolines via gold-catalyzed cascade cyclizations and enantioselective reactions

图式22 金催化吲哚-炔胺底物的分子内串联环化反应
Scheme 22 Gold-catalyzed intramolecular tandem cyclization of indole-ynamides

图式23 Ugi/Au(Ⅰ)催化的多米诺环化反应合成螺-吲哚啉类化合物
Scheme 23 Ugi/gold-catalyzed diastereoselective domino cyclization

图式24 经炔丙基重排的金催化2-炔基-N-炔丙基苯胺的串联环化反应
Scheme 24 Gold-catalyzed cascade cyclization of 2-alkynyl-N-propargylanilines by rearrangement of a propargyl group

图式26 金催化吲哚与联烯酰胺分子间去芳构化反应
Scheme 26 Taming gold(Ⅰ)-counterion interplay in the dearomatization of indoles with allenamides

图式27 不对称金催化吲哚与联烯酰胺分子间串联去芳构化反应
Scheme 27 Enantioselective gold-catalyzed cascade construction of pyrroloindoline derivatives

图式28 不对称金催化吲哚与联烯酰胺[2+2]环加成反应
Scheme 28 Enantioselective gold-catalyzed dearomative [2+2]-cycloaddition between indoles and allenamides

图式29 金催化烯基吲哚与联烯酰胺分子间[4+2]环加成反应
Scheme 29 Gold-catalyzed intermolecular [4+2]-cycloaddition of vinyl indoles and N-allenamides
-


 下载:
下载:






























 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 57
- 文章访问数: 2530
- HTML全文浏览量: 557
 下载:
下载:
