 图 1
采用一步法, 不同时间的样品吸收光谱图 (A) 和荧光光谱图 (B)
Figure 1.
The evolution of (A) absorption spectra and (B) PL spectra of the samples with reaction time by one step synthesis
图 1
采用一步法, 不同时间的样品吸收光谱图 (A) 和荧光光谱图 (B)
Figure 1.
The evolution of (A) absorption spectra and (B) PL spectra of the samples with reaction time by one step synthesis
引用本文:
黄璐, 李志春, 黄寿强, PeterReiss, 李良. 持续注射法合成InPZnS/ZnS核壳结构量子点的研究[J]. 化学学报,
2017, 75(3): 300-306.
doi:
10.6023/A16100543

Citation: Huang Lu, Li Zhichun, Huang Shouqiang, Peter Reiss, Li Liang. Synthesis of InPZnS/ZnS Quantum Dots by Continuous Injection of Phosphorus Precursor[J]. Acta Chimica Sinica, 2017, 75(3): 300-306. doi: 10.6023/A16100543
Citation: Huang Lu, Li Zhichun, Huang Shouqiang, Peter Reiss, Li Liang. Synthesis of InPZnS/ZnS Quantum Dots by Continuous Injection of Phosphorus Precursor[J]. Acta Chimica Sinica, 2017, 75(3): 300-306. doi: 10.6023/A16100543
持续注射法合成InPZnS/ZnS核壳结构量子点的研究
摘要:
高质量、大尺寸的磷化铟量子点(InP quantum dots)的制备对其在生物荧光标记等领域的应用有重大的意义.提出一种持续注射法合成InPZnS/ZnS核壳结构量子点.首先将磷化锌和硫酸反应生成的磷化氢持续注入高温铟前体溶液中,并在反应开始加入适量的锌和硫前体,可制得合金结构的InPZnS核,其量子产率达9%.该方法无需加锌回流处理,可直接升温包覆ZnS壳层,成功制备出发光波长至680 nm且量子产率接近50%的InPZnS/ZnS核壳结构量子点.同时系统地研究了反应物配比对量子点粒径的影响规律,结果表明:通过调控反应物配比可合成不同粒径的InP量子点,其光谱范围几乎可覆盖整个可见光区,甚至到近红外区.通过吸收光谱、荧光光谱、透射电子显微镜、X射线衍射仪和X射线能谱仪考察了所制备量子点的光学特性和形貌结构.最后,采用其它磷前体探究了此方法的通用性,通过注射泵将三(二乙氨基)膦持续注入到高温铟前体溶液中,同样制备出较大尺寸的InP量子点,其发光波长至710 nm.
-
关键词:
- InPZnS/ZnS
- / 核壳型量子点
- / 量子产率
- / 光学特性
- / 持续注射法
English
Synthesis of InPZnS/ZnS Quantum Dots by Continuous Injection of Phosphorus Precursor
Abstract:
InP quantum dots (QDs) are regarded as the most desirable candidate to replace the role of CdSe QDs in the applications of bio-labeling, LEDs, solar cells, etc, because InP is more environmentally friendly compared to Cd based QDs, and could also offer a tunable emission from blue to near-infrared. Nevertheless, the studies and applications of InP QDs are rather sparse in comparison with CdSe QDs, which are principally caused by significant difficulties in its synthesis. In this report, we developed a novel method for the synthesis of InPZnS/ZnS QDs by using zinc phosphide as phosphorus precursor, and the zinc and sulfur precursors were also added at the start of reaction, which allows the continuous injection of phosphine gas into the reaction, resulting in high quality InPZnS/ZnS quantum dots with emission up to 680 nm. The core synthesis and shell coating were separated by controlling the reaction temperature. During the first 30 minutes, the temperature of reaction solution was kept at 250℃ to grow the InPZnS core QDs. Then, the coating of ZnS shell was happened and kept about 1 hour to guarantee the complete decomposition of 1-dodecanethiol (DDT) after the reaction temperature was increased to 300℃. The biggest advantage of this synthetic method is the tunable emission region from blue to near-infrared. The effects of reaction parameters were systematically investigated. We observed that the molar ratio of In:myristic acid (MA) and that of In:Zn (S) had significant influences on the size of the InP QDs. The structure of InPZnS/ZnS QDs was confirmed by transmission electron microscope (TEM), X-ray powder diffraction (XRD), and energy dispersive X-ray analyzer (EDX). TEM characterization indicated the final core/shell InPZnS/ZnS QDs were good monodispersity with an average size of 7 nm. Furthermore, we investigated the versatility of this method by using other phosphorus precursor. The injection pump leaded to a continuous supply of phosphorus precursor on a timescale and reacted with indium precursor to form InP QDs. The final sample showed an emission at 710 nm. The present method gives access to larger sized InP QDs, making it prosperous for applications in biological labeling.
-
Key words:
- InPZnS/ZnS
- / core/shell QDs
- / quantum yield
- / optical property
- / continuous injection
-
1 引言
量子点 (Quantum dots), 又称为半导体纳米晶体 (Semiconductor nanocrystal).从20世纪80年代被发现以来[1], 由于其优异的光致发光性能和独特的带隙可调性而备受科研界和工业界的关注, 目前量子点已被广泛应用于显示[2]、照明[3]和生物荧光标记[4, 5]等领域.近年来, 研究者对以CdSe为代表的ⅡB~Ⅵ族量子点进行了广泛的研究, 其合成技术已较为成熟.但随着研究的深入, 人们发现Cd元素的生物毒性限制了Cd系量子点在细胞成像、免疫分析、生物传感器等领域的应用[6].与之相比, 以InP为代表的Ⅲ~Ⅴ族量子点毒性较低, 具有优异的生物相容性, 通过调控量子点的粒径, 其光谱范围可基本覆盖整个可见光区, 甚至到近红外区[7].然而, 由于InP量子点难以制备, 目前国内外关于InP量子点的研究与应用仍较少.
热注射法是一种常用的合成高质量、单分散量子点的有效方法, 已成功应用于CdSe等ⅡB~Ⅵ族量子点的合成.目前, 制备InP量子点也普遍采用这种方法, 将常温磷前体快速注射到高温铟前体溶液中, 使其成核并生长.此方法一次性注入大量活性极高的磷前体, 根据经典成核理论, 前体的浓度越高, 形成的晶种越多, 导致最终合成的量子点的平均粒径越小[8].因此, 采用此方法一般难以合成大尺寸、发射光谱范围达到近红外区的InP量子点.由于P元素在晶体表面易形成大量的悬挂键[9, 10], 致使合成的InP量子点量子产率低 (QY<1%)、稳定性差.通过外延生长法在量子点表面包覆另一种半导体材料形成核壳结构, 能够有效地钝化量子点表面, 从而大幅度提高其量子产率和稳定性[11].但因InP纳米晶表面极易被氧化生成氧化铟, 致使难以在其表面直接包覆ZnS, 所以一般都会在包覆前对InP裸核进行前处理[8].将硬脂酸锌加入到预先制备好的InP溶液中, 在270 ℃高温回流1~4 h后形成InPZn复合物, 再向溶液中加入硫前体反应生成ZnS壳层, 此方法可合成出较高质量的InP/ZnS核壳结构量子点.此外, Reiss等[12]提出了一锅法合成InP/ZnS量子点, 量子产率高达50%~70%, 且具有较好的稳定性.该方法无需前处理, 即可成功制得高质量的量子点, 主要是由于在合成InP核时就加入了锌和硫前体, 形成了一种合金结构的InPZnS核[13, 14], 有效修复了晶体表面缺陷, 无需加锌回流就可直接包覆ZnS.
上述合成方法均使用三 (三甲硅烷基) 磷[P (TMS)3]作为磷源, 但其极易燃且价格昂贵, 不适于InP量子点的大批量合成及应用.为此, Li等[15]用磷化钙 (Ca3P2) 和盐酸反应生成的磷化氢作为磷前体, 合成的InP量子点量子产率为22%. Zan等[16]使用磷化锌 (Zn3P2) 代替磷化钙和盐酸反应生成磷化氢, 制备了高质量的InP/ZnS核壳结构量子点.以上两种方法大幅度降低了制备成本, 但合成的InP核量子产率均较低, 且在包覆前需加锌回流4~9 h.
综合上述几种制备方法, 工作提出了一种新型的InP/ZnS核壳结构量子点合成方法.将磷化锌和硫酸反应生成的磷化氢气体持续通入高温铟前体溶液中, 并在反应开始加入适量的锌和硫前体, 制得一种合金结构的InPZnS核, 其量子产率提高到9%.不需加锌回流处理, 可直接升温包覆ZnS壳层, 成功制得了高荧光量子产率的核壳型量子点.我们系统地研究了不同实验条件对量子产率的影响, 在优化的条件下合成出量子产率约50%的InPZnS/ZnS量子点, 同时通过调控配比制得粒径较大的量子点, 其荧光光谱范围能覆盖可见光区及近红外区, 有利于其在生物医学领域的应用.
2 结果与讨论
2.1 InPZnS/ZnS核壳型量子点的制备
在不同的反应温度下分阶段进行InPZnS核的制备和ZnS壳层的包覆, 包覆过程又分一步法和两步法.一步法在反应开始就将硬脂酸锌和十二烷基硫醇 (DDT) 一次性加入.两步法即分两步加入锌和硫前体, 核制备阶段只加入少量的硬脂酸锌和DDT, 后续包覆开始时再补充加入适量.首先合成InPZnS核, 反应温度控制在250 ℃, 反应时间为30 min. 图 1为采用一步法, 不同反应时间的样品吸收光谱图 (图 1A) 和荧光光谱图 (图 1B).
图 1
采用一步法, 不同时间的样品吸收光谱图 (A) 和荧光光谱图 (B)
Figure 1.
The evolution of (A) absorption spectra and (B) PL spectra of the samples with reaction time by one step synthesis
由图 1B可见, 前20 min, 随着反应的进行荧光峰强度逐渐增强, 峰的位置持续红移, 由552 nm移动到565 nm, 半峰宽从72 nm变为62 nm (如表 1所示), 说明量子点在快速生长的同时其粒径分布也较好.反应进行到20 min后, 由于Zn3P2的不断消耗, PH3的生成速率逐渐减小, 量子点并没有随着时间的延长而继续生长, 激子吸收峰的位置基本保持不变 (如图 1A所示), 说明量子点已停止生长. DDT的分解温度为280 ℃, 但在实际混合液中, 250 ℃时DDT已经开始缓慢分解, 有利于制备高质量的InP量子点.反应中DDT分解释放出S2-与溶液中的Zn结合形成了合金结构的InPZnS核, 部分消除量子点表面的悬挂键.由此, 量子点荧光强度增强, 荧光量子产率达9%左右, 较之不加锌和硫前体直接制得的纯InP核的量子产率 (QY<1%) 高很多. 30 min后将温度升至300 ℃, 反应到35 min量子产率显著提高, 从9%增加到30%(如表 1所示), 表明量子点表面已经包覆ZnS壳层.随着包覆反应的进行, 激子吸收峰和荧光峰的位置持续红移, 说明量子点的粒径逐渐增大, 包覆的壳层逐渐变厚.反应1 h后, 量子点荧光峰的位置在577 nm处, 荧光强度大幅增强, 量子产率提高至48.4%, 半峰宽为58.8 nm, 表明包覆ZnS壳层效果显著, 充分钝化了量子点的表面, 有效减少了表面缺陷, 在提高量子点量子产率的同时也能控制好其粒径分布[17].
 表 1
不同反应时间的样品荧光峰的位置、量子产率和荧光峰半峰宽值
Table 1.
The quantum yield (QY) and full-width at half-maximum (FWHM) of the as prepared samples with reaction time
表 1
不同反应时间的样品荧光峰的位置、量子产率和荧光峰半峰宽值
Table 1.
The quantum yield (QY) and full-width at half-maximum (FWHM) of the as prepared samples with reaction time
Time 5 min 10 min 20 min 30 min 35 min 40 min 50 min 60 min 70 min 90 min Peak/nm 552 559 565 564 563 567 569 570 572 577 QYs/% 0.70 2.20 6.00 9.20 30.0 31.0 32.0 37.0 44.0 48.4 FWHM/nm 72.0 67.2 62.0 60.0 58.0 58.8 58.0 59.4 58.8 58.8 表 1 不同反应时间的样品荧光峰的位置、量子产率和荧光峰半峰宽值
Table 1. The quantum yield (QY) and full-width at half-maximum (FWHM) of the as prepared samples with reaction time2.2 反应物配比的影响
此制备方法的优势之一是能通过调控反应物的配比, 合成出不同粒径、不同发射波长的InP量子点, 其光谱范围几乎可覆盖整个可见光及近红外区.本文系统地研究了不同反应条件对InP量子点粒径的影响.首先研究了磷前体 (Zn3P2) 的用量对量子点粒径的影响, 如图 2所示, 改变In:P物质的量比, 量子点的激子吸收峰的位置无明显移动, 表明In:P物质的量比 (<1) 对量子点的粒径影响不大.
 图 2
不同的In:P物质的量比下, 不同反应时间的样品吸收光谱图 (采用一步法制备样品, n(In):n(MA):n(Zn):n(S)=1:3:1:1)
Figure 2.
The evolution of absorption spectra of the samples synthesized at different In:P molar ratios with reaction time by one step synthesis. [n(In):n(MA):n(Zn):n(S)=1:3:1:1]
图 2
不同的In:P物质的量比下, 不同反应时间的样品吸收光谱图 (采用一步法制备样品, n(In):n(MA):n(Zn):n(S)=1:3:1:1)
Figure 2.
The evolution of absorption spectra of the samples synthesized at different In:P molar ratios with reaction time by one step synthesis. [n(In):n(MA):n(Zn):n(S)=1:3:1:1]
根据相关文献报道[7, 12, 18], 反应体系中In前体的活性影响InP量子点的粒径, 而前者又与体系中加入的肉豆蔻酸 (MA) 量有关, 因此, 通过改变In:MA的物质的量比可以调控InP量子点的粒径大小.如图 3所示, 当溶液中MA浓度较低时[n(In):n(MA)=1:1.5或1:3], 反应进行到10 min左右, 激子吸收峰的位置基本稳定, 保持在505~510 nm处, 合成的InP量子点粒径较小.这是由于MA浓度较低, 不能有效抑制In前体活性, In和P前体快速反应生成大量的InP晶核, 短时间内In前体被快速消耗, 影响了量子点的后续生长, 使得最终生成的量子点粒径较小.增加MA的浓度[n(In):n(MA)=1:5], 产物激子吸收峰的位置呈现大幅度红移, 最终位于580 nm处, 这是由于随着MA浓度增大, In反应活性受到抑制, 成核阶段生成的晶核减少, 避免了短时间内In前体的快速消耗, 持续通入的PH3气体与混合液中剩余的In继续反应, 不断生成InP沉积于反应体系已有的晶核表面, 使InP量子点继续长大, 因此可得到较大粒径的InP量子点[8].
 图 3
不同的In:MA物质的量比下, 不同反应时间的样品吸收光谱图 (采用一步法制备样品, n(In):n(P):n(Zn):n(S)=1:6:1:1)
Figure 3.
The evolution of absorption spectra of the samples synthesized at different In:MA molar ratios with reaction time by one step synthesis. [n(In):n(P):n(Zn):n(S)=1:6:1:1]
图 3
不同的In:MA物质的量比下, 不同反应时间的样品吸收光谱图 (采用一步法制备样品, n(In):n(P):n(Zn):n(S)=1:6:1:1)
Figure 3.
The evolution of absorption spectra of the samples synthesized at different In:MA molar ratios with reaction time by one step synthesis. [n(In):n(P):n(Zn):n(S)=1:6:1:1]
将硬脂酸锌和DDT预先加入到铟前体溶液中, 250 ℃高温下反应30 min, 最终生成了一种合金结构的InPZnS核, 其荧光量子效率高于纯InP纳米晶核.但实验中我们发现若预先加入过多的硬脂酸锌和DDT会影响InP纳米晶的进一步生长, 所以制备较大粒径的量子点需减少锌和硫前体的添加量.由图 4A可见, 随着反应最初锌和硫前体加入量的减少, 量子点的激子吸收峰的位置持续红移, 说明量子点的粒径逐渐增大, 证实了减少锌和硫前体的量有利于InP纳米晶的生长, 但若锌和硫前体的量过少, 包覆在InPZnS纳米晶核表面的ZnS层将不足以充分钝化量子点表面.
 图 4
(A) 改变反应最初加入的In:Zn (S) 物质的量比, 合成的不同InPZnS核的吸收光谱图[n(In):n(P):n(MA)=1:6:3];采用两步法包覆, 不同反应时间的样品吸收光谱图 (B) 和荧光光谱图 (C)
Figure 4.
(A) The absorption spectra of the samples synthesized at different In:Zn (S) molar ratios with reaction time. [n(In):n(P):n(MA)=1:6:3]. The evolution of absorption (B) and PL (C) spectra of the samples synthesized by two-step synthesis with reaction time
图 4
(A) 改变反应最初加入的In:Zn (S) 物质的量比, 合成的不同InPZnS核的吸收光谱图[n(In):n(P):n(MA)=1:6:3];采用两步法包覆, 不同反应时间的样品吸收光谱图 (B) 和荧光光谱图 (C)
Figure 4.
(A) The absorption spectra of the samples synthesized at different In:Zn (S) molar ratios with reaction time. [n(In):n(P):n(MA)=1:6:3]. The evolution of absorption (B) and PL (C) spectra of the samples synthesized by two-step synthesis with reaction time
为避免这一现象的发生, 在InPZnS纳米晶核制备完成后, 可快速再注入适量的硬脂酸锌和DDT以满足后续包覆过程的需要.文中我们称之为两步法制备InPZnS/ZnS核壳结构量子点, 即分两步加入锌和硫前体, 核制备阶段只加入少量的硬脂酸锌和DDT [n(In):n[Zn (S)]=1:0.15, n(In)=0.1 mmol], 包覆开始时再加入1 mmol硬脂酸锌和1 mmol DDT.由图 4B可见, 第一阶段反应30 min生成的InPZnS核的激子吸收峰在620 nm处, 表明制得了较大粒径的InPZnS纳米晶核, 由于采用两步法包覆时又加入了一定量的硬脂酸锌和DDT, 包覆的ZnS壳层变厚, 充分钝化了量子点表面, 量子点荧光强度大幅度增加, 包覆ZnS (1 h) 后荧光峰的位置位于680 nm处, 成功合成出较大粒径的InPZnS/ZnS核壳结构量子点.
通过改变反应物的配比, 在优化的条件下, 我们合成了高质量、不同粒径的InPZnS/ZnS核壳结构量子点. 图 5A为不同粒径的InPZnS/ZnS核壳量子点在紫外灯照射下的图像, 产物发射光可从绿色到深红色, 且量子产率约50%. 图 5B为各颜色相对应的InPZnS/ZnS核壳量子点的吸收和荧光光谱图, 由图可知其荧光光谱范围覆盖550~680 nm.
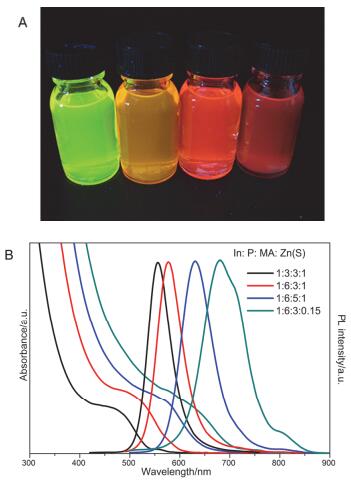 图 5
(A) 不同粒径的InPZnS/ZnS核壳量子点在紫外灯照射下的图像; (B) 不同粒径的InPZnS/ZnS量子点吸收和荧光光谱图
Figure 5.
(A) The photograph of different sized InPZnS/ZnS QDs under UV light. (B) The PL (λex 450 nm) and absorption spectra of those different sized InPZnS/ZnS QDs
图 5
(A) 不同粒径的InPZnS/ZnS核壳量子点在紫外灯照射下的图像; (B) 不同粒径的InPZnS/ZnS量子点吸收和荧光光谱图
Figure 5.
(A) The photograph of different sized InPZnS/ZnS QDs under UV light. (B) The PL (λex 450 nm) and absorption spectra of those different sized InPZnS/ZnS QDs
2.3 InPZnS/ZnS核壳型量子点的结构
透射电子显微镜 (TEM) 表征结果, 如图 6所示.成核阶段, 250 ℃高温下反应30 min形成了合金结构的InPZnS核, 其平均粒径为2.9 nm (图 6A), 采用两步法合成的InPZnS/ZnS量子点的平均粒径为7 nm (图 6C), 明显大于InPZnS核粒径, 成功于InPZnS核表面包覆ZnS层形成了核壳结构量子点.采用一步法合成出的InPZnS/ZnS量子点的平均粒径相对小些, 约为4 nm (图 6B), 此方法将锌和硫前体于反应开始时一次性加入[n(In):n[Zn (S)]=1:1], 浓度较高阻止了InPZnS核的生长, 而后续包覆没有补充Zn (S) 前体, 所以制得的InPZnS/ZnS量子点的粒径相对较小, 但仍大于InPZnS纳米晶核的平均粒径.
 图 6
(A) InPZnS核的透射电镜图 (λem=650 nm); (B) 一步法合成的InPZnS/ZnS量子点的透射电镜图 (λem=577 nm, In:P:MA:Zn:S=1:6:3:1:1); (C) 两步法合成的InPZnS/ZnS量子点的透射电镜图 (λem=680 nm, 成核过程: In:P:MA:Zn:S=1:6:3:0.15:0.15, In=0.1 mmol; 包覆过程:再注入1 mmol的Zn (St)2和1 mmol DDT)
Figure 6.
(A) TEM images of the as-prepared InPZnS QDs. (λem=650 nm). (B) TEM images of InPZnS/ZnS QDs by one step synthesis. (λem=577 nm, In:P:MA:Zn:S=1:6:3:1:1) (C) TEM images of InPZnS/ZnS QDs by two-step synthesis. (λem=680 nm, Core synthesis: In:P:MA:Zn:S=1:6:3:0.15:0.15, In=0.1 mmol, Shell synthesis: 1 mmol Zn (st)2, 1 mmol DDT and 5 mL ODE)
图 6
(A) InPZnS核的透射电镜图 (λem=650 nm); (B) 一步法合成的InPZnS/ZnS量子点的透射电镜图 (λem=577 nm, In:P:MA:Zn:S=1:6:3:1:1); (C) 两步法合成的InPZnS/ZnS量子点的透射电镜图 (λem=680 nm, 成核过程: In:P:MA:Zn:S=1:6:3:0.15:0.15, In=0.1 mmol; 包覆过程:再注入1 mmol的Zn (St)2和1 mmol DDT)
Figure 6.
(A) TEM images of the as-prepared InPZnS QDs. (λem=650 nm). (B) TEM images of InPZnS/ZnS QDs by one step synthesis. (λem=577 nm, In:P:MA:Zn:S=1:6:3:1:1) (C) TEM images of InPZnS/ZnS QDs by two-step synthesis. (λem=680 nm, Core synthesis: In:P:MA:Zn:S=1:6:3:0.15:0.15, In=0.1 mmol, Shell synthesis: 1 mmol Zn (st)2, 1 mmol DDT and 5 mL ODE)
X射线粉末衍射光谱图 (XRD) 进一步证明了量子点核壳结构的形成.如图 7所示, InP纳米晶核、InPZnS纳米晶核与包覆了ZnS的核壳量子点均属于立方晶形 (闪锌矿).加热到300 ℃包覆ZnS后, 无论是采用一步法还是两步法合成的InPZnS/ZnS量子点衍射峰的位置均向ZnS的标准图谱偏移, 但采用两步法包覆的ZnS层较厚, 形成的InPZnS/ZnS量子点基本呈现ZnS的衍射峰, 证实了ZnS壳层的存在.
 图 7
(A) InP纳米晶核的X射线粉末衍射光谱图 (XRD); (B) InPZnS纳米晶核的XRD图; (C) 一步法制备的InPZnS/ZnS核壳量子点的XRD图; (D) 采用两步法制备的InPZnS/ZnS核壳量子点的XRD图
Figure 7.
(A) Powder X-ray Diffractograms of InP QDs. (B) Powder X-ray Diffractograms of InPZnS QDs. (C) Powder X-ray Diffractograms of InPZnS/ZnS QDs by one step synthesis. [n(In):n(Zn)=1:1] (D) Powder X-ray Diffractograms of InPZnS/ZnS QDs by two-step synthesis. [n(In):n(Zn)=1:1] For comparison the diffraction patterns of bulk cubic InP (triangles) and ZnS (squares) are displayed.
图 7
(A) InP纳米晶核的X射线粉末衍射光谱图 (XRD); (B) InPZnS纳米晶核的XRD图; (C) 一步法制备的InPZnS/ZnS核壳量子点的XRD图; (D) 采用两步法制备的InPZnS/ZnS核壳量子点的XRD图
Figure 7.
(A) Powder X-ray Diffractograms of InP QDs. (B) Powder X-ray Diffractograms of InPZnS QDs. (C) Powder X-ray Diffractograms of InPZnS/ZnS QDs by one step synthesis. [n(In):n(Zn)=1:1] (D) Powder X-ray Diffractograms of InPZnS/ZnS QDs by two-step synthesis. [n(In):n(Zn)=1:1] For comparison the diffraction patterns of bulk cubic InP (triangles) and ZnS (squares) are displayed.
表 2为X射线能谱图 (EDX) 中不同包覆时间的InPZnS/ZnS量子点的各种元素的百分比.成核阶段反应温度为250 ℃, 通入高活性的PH3气体与铟前体溶液反应生成InPZnS纳米晶核.在这个阶段, 由于温度较低, 仅分解了少量的DDT, 进入包覆阶段将温度升高到300 ℃, DDT的分解速度变快, 随着反应进行, 锌和硫在量子点中的含量不断增加 (如表 2所示), 采用一步法包覆1 h后形成的InPZnS/ZnS核壳量子点中锌和铟的物质的量比约为0.6:1.而两步法制备的InPZnS/ZnS核壳量子点中锌和硫的含量大幅提高, 锌和铟的物质的量比增大到4.5:1左右, 表明与一步法包覆的ZnS壳层相比两步法的明显较厚.
表 2
X射线能谱图 (EDX) 中不同包覆时间的InPZnS/ZnS量子点的各种元素的百分比: (A) 采用一步法, 各元素物质的量比为n(In):n(P):n(MA):n(Zn):n(S)=1:1:3:1:1时, 30, 40, 60, 90 min的样品; (B) 两步法制备的样品:成核过程, 各元素物质的量比为n(In):n(P):n(MA):n(Zn):n(S)=1:1:6:0.15:0.15; 30 min后, 补充加入一定量的锌和硫前体进行ZnS包覆. [n(In):n(Zn):n(S)=1:10:10]
Table 2.
The ratio of these elements of samples with different shell thickness from the EDX spectra (JEOL JSM-840A SEM equipped with an Oxford Instruments energy dispersive X-ray analyzer): (A) synthesized by one step synthesis: n(In):n(P):n(MA):n(Zn):n(S)=1:1:3:1:1, 30, 40, 60, 90 min; (B) synthesized by two-step synthesis: Core synthesis, n(In):n(P):n(MA):n(Zn):n(S)=1:1:6:0.15:0.15, 30 min, and coated ZnS shell by adding plus Zinc and sulfur precursors. [n(In):n(Zn):n(S)=1:10:10]
Elt/A% A30 min A40 min A60 min A90 min B90 min P 41.16 40.48 33.05 32.42 8.67 S 10.77 12.18 16.89 22.35 47.39 Zn 10.19 12.92 17.74 16.68 35.87 In 37.88 34.42 32.32 28.55 8.07 Total 100.00 100.00 100.00 100.00 100.00 表 2 X射线能谱图 (EDX) 中不同包覆时间的InPZnS/ZnS量子点的各种元素的百分比: (A) 采用一步法, 各元素物质的量比为n(In):n(P):n(MA):n(Zn):n(S)=1:1:3:1:1时, 30, 40, 60, 90 min的样品; (B) 两步法制备的样品:成核过程, 各元素物质的量比为n(In):n(P):n(MA):n(Zn):n(S)=1:1:6:0.15:0.15; 30 min后, 补充加入一定量的锌和硫前体进行ZnS包覆. [n(In):n(Zn):n(S)=1:10:10]
Table 2. The ratio of these elements of samples with different shell thickness from the EDX spectra (JEOL JSM-840A SEM equipped with an Oxford Instruments energy dispersive X-ray analyzer): (A) synthesized by one step synthesis: n(In):n(P):n(MA):n(Zn):n(S)=1:1:3:1:1, 30, 40, 60, 90 min; (B) synthesized by two-step synthesis: Core synthesis, n(In):n(P):n(MA):n(Zn):n(S)=1:1:6:0.15:0.15, 30 min, and coated ZnS shell by adding plus Zinc and sulfur precursors. [n(In):n(Zn):n(S)=1:10:10]2.4 持续注射法的通用性
文中采用稳定的Ar气流将生成的磷化氢气体通入高温In前体溶液中, 并在反应开始加入适量的锌和硫前体, 通过调节反应物的配比合成了高质量、大尺寸的InPZnS/ZnS核壳量子点.此方法实质为持续注射法, 在核制备阶段, 通过稳定的Ar气流将磷化锌和硫酸反应生成的PH3气体持续地通入In前体溶液中, 避免了短时间内In前体和P前体的快速消耗, 延长了量子点的成核和生长时间, 由此合成的InPZnS纳米晶核较大.将此方法用于其它磷前体 (三 (二乙氨基) 膦) 合成InP量子点, 采用注射泵, 将该P前体持续注入到In前体溶液中, 250 ℃高温下反应30 min, 由图 8A可见, 30 min后激子吸收峰的位置在660 nm处, 制得了较大粒径的InPZnS纳米晶核.采用两步法包覆ZnS (1 h) 后荧光峰的位置在710 nm处 (图 8B所示), 合成了较大粒径的InPZnS/ZnS核壳结构量子点, 验证了变换磷前体, 持续注射法仍能合成出大尺寸的InP量子点, 证实了该方法的通用性.
 图 8
改变磷前体, 采用两步法合成的不同时间的样品吸收光谱图 (A) 和荧光光谱图 (B)
Figure 8.
The evolution of (A) absorption spectra and (B) PL spectra of the samples with reaction time by two-step synthesis
图 8
改变磷前体, 采用两步法合成的不同时间的样品吸收光谱图 (A) 和荧光光谱图 (B)
Figure 8.
The evolution of (A) absorption spectra and (B) PL spectra of the samples with reaction time by two-step synthesis
3 结论
本工作采用磷化锌和硫酸反应生成的磷化氢作为磷源, 通过持续注射法合成InP/ZnS核壳结构量子点, 该方法在反应前将锌和硫前体加入In前体溶液中, 实质上合成了一种合金结构的InPZnS/ZnS核壳量子点.在优化的反应条件下, 成功制备出高量子产率、荧光峰半峰宽较窄的核壳结构量子点.此外, 与普通手动注射法相比, 持续注射法可利用自动注射泵进行持续注射, 使得实验的重复性和操作性更好.通过调控各反应参数, 可以合成出大尺寸、高质量及荧光光谱范围覆盖到近红外区的量子点, 极大地促进了低毒性的InP量子点在生物医学等领域的应用.
4 实验部分
4.1 主要试剂
醋酸铟 (99.99%)、十二烷基硫醇 (DDT, 99.99%)、硬脂酸锌 (Zn (St)2, 99%)、1-十八烯 (ODE, 90%)、磷化锌 (Zn3P2)、三 (二乙氨基) 膦均购自Sigma公司, 硫酸购买于国药上海试剂公司.以上药品均在未经过任何预处理下直接使用.
4.2 InPZnS/ZnS核壳型量子点的制备
InPZnS核的合成:在手套箱中将0.1 mmol的醋酸铟, 0.1 mmol的硬脂酸锌, 0.1 mmol的十二烷基硫醇以及8 mL的1-十八烯加入到三颈烧瓶中 (A), 将25.8 mg (0.1 mmol) 的磷化锌加入另一个三颈瓶中 (B), 反应装置如图 9所示.
 图 9
合成InPZnS/ZnS核壳量子点的实验装置图
Figure 9.
Experimental Setup for the Synthesis of InPZnS/ZnS QDs
图 9
合成InPZnS/ZnS核壳量子点的实验装置图
Figure 9.
Experimental Setup for the Synthesis of InPZnS/ZnS QDs
烧瓶A在100~120 ℃下反复抽真空1 h直至浑浊的混合液变为无色透明液体, 然后在Ar保护下升温至250 ℃.随后将2 mL浓度为1 mol/L的硫酸注入烧瓶B中, 与磷化锌反应生成磷化氢气体, 通过稳定的Ar气流将生成的磷化氢气体引入到烧瓶A中, 与In前体溶液反应生成InPZnS核, 溶液由无色逐渐变为深红色 (5~10 min).
包覆ZnS壳层:此过程分一步法和两步法. 250 ℃反应30 min后, 直接将烧瓶A升温至300 ℃进行ZnS壳层的包覆, 文中称之为一步法; 两步法即分两步加入锌和硫前体, 成核阶段只加入少量的硬脂酸锌和DDT, 各组分物质的量比为n(In):n(P):n(MA):n(Zn):n(S)=1:6:3:0.15:0.15 [n(In)=0.1 mmol], 后续升温至300 ℃开始包覆时再补充加入1 mmol的硬脂酸锌和1 mmol DDT.
待制得的量子点溶液冷却至室温后, 加入适量的丙酮和甲醇混合液 (1:1), 离心, 将所得沉淀溶解在甲苯、正己烷、氯仿等有机溶剂中.
4.3 量子点的表征
量子点的吸收光谱测量采用U-3900紫外-可见光谱仪 (日本日立公司), 荧光光谱测量采用F-380荧光光谱仪 (港东科技有限公司), 激发波长为450 nm. X射线衍射晶体结构分析采用飞利浦公司的X’PERT型X射线衍射仪 (Co靶Kα辐射, 激发, λ=1.789 ).采用透射电子显微镜 (JEOL2010, 工作电压在200 kV) 来观测量子点的尺寸大小和晶体结构.
-
-
[1]
Bera, D.; Qian, L.; Tseng, T. K.; Holloway, P. H. Materials 2010, 3, 2260. doi: 10.3390/ma3042260
-
[2]
Bourzac, K. Nature 2013, 493, 283. doi: 10.1038/493283a
-
[3]
Zhang, Y.; Xie, C.; Su, H. P.; Liu, J.; Pickering, S.; Wang, Y. Q.; Yu, W. W.; Wang, J. K.; Wang, Y. D.; Hahm, J.; Dellas, N.; Mohney, S. E.; Xu, J. Nano Lett. 2011, 11, 329. doi: 10.1021/nl1021442
-
[4]
Michalet, X.; Pinaud, F. F.; Bentolila, L. A.; Tsay, J. M.; Doose, S.; Li, J. J.; Sundaresan, G.; Wu, A. M.; Gambhir, S. S.; Weiss, S. Science 2005, 307, 538. doi: 10.1126/science.1104274
-
[5]
Li, L.; Daou, J.; Texier, I.; Chi, T. T. K.; Liem, N. Q.; Reiss, P. Chem. Mater. 2009, 21, 2422. doi: 10.1021/cm900103b
-
[6]
Chen, F. Q.; Gerion, D. Nano Lett. 2004, 4, 1827. doi: 10.1021/nl049170q
-
[7]
Xie, R. G.; Battaglia, D.; Peng, X. G. J. Am. Chem. Soc. 2007, 129, 15432. doi: 10.1021/ja076363h
-
[8]
Tamang, S.; Lincheneau, C.; Hermans, Y.; Jeong, S.; Reiss, P. Chem. Mater. 2016, 28, 2491. doi: 10.1021/acs.chemmater.5b05044
-
[9]
Adam, S.; Talapin, D.; Borchert, H.; Lobo, A.; McGinley, C.; De Castro, A.; Hasse, M.; Weller, H.; Möller, T. J. Chem. Phys. 2005, 123, 084706. doi: 10.1063/1.2004901
-
[10]
Lovingood, D. D.; Strouse, G. F. Nano Lett. 2008, 8, 3394. doi: 10.1021/nl802075j
-
[11]
李谦, 张腾, 古宏伟, 丁发柱, 屈飞, 彭星煜, 王洪艳, 吴战鹏, 化学学报, 2013, 71, 929. doi: 10.6023/A13010052Li, Q.; Zhang, T.; Gu, H. W.; Ding, F. Z.; Qu, F.; Peng, X. Y.; Wang, H. Y.; Wu, Z. P. Acta Chim. Sinica 2013, 71, 929. doi: 10.6023/A13010052
-
[12]
Li, L.; Reiss, P. J. Am. Chem. Soc. 2008, 130, 11588. doi: 10.1021/ja803687e
-
[13]
Kim, T. H.; Kim, S. W.; Kang, M. J.; Kim, S. W. J. Phys. Chem. Lett. 2012, 3, 214. doi: 10.1021/jz201605d
-
[14]
Altıntas, Y.; Talpur, M. Y.; Ünlu, M.; Mutlugün, E. J. Phys. Chem. C 2016, 120, 7885. doi: 10.1021/acs.jpcc.6b01977
-
[15]
Li, L.; Protière, M.; Reiss, P. Chem. Mater. 2008, 20, 2621. doi: 10.1021/cm7035579
-
[16]
Zan, F.; Ren, J. C. J. Mater. Chem. 2012, 22, 1794. doi: 10.1039/C1JM13982G
-
[17]
陈美华, 潘峥, 尹月锋, 刘洁, 刘梦媛, 贾紫君, 梁桂杰, 化学学报, 2016, 74, 330. doi: 10.6023/A15120785Chen, M. H.; Pan, Z.; Yin, Y. F.; Liu, J.; Liu, M. Y.; Jia, Z. J.; Liang, G. J. Acta Chim. Sinica 2016, 74, 330. doi: 10.6023/A15120785
-
[18]
Xu, S.; Ziegler, J.; Nann, T. J. Mater. Chem. 2008, 18, 2653. doi: 10.1039/b803263g
-
[1]
-

图 1 采用一步法, 不同时间的样品吸收光谱图 (A) 和荧光光谱图 (B)
Figure 1 The evolution of (A) absorption spectra and (B) PL spectra of the samples with reaction time by one step synthesis

图 2 不同的In:P物质的量比下, 不同反应时间的样品吸收光谱图 (采用一步法制备样品, n(In):n(MA):n(Zn):n(S)=1:3:1:1)
Figure 2 The evolution of absorption spectra of the samples synthesized at different In:P molar ratios with reaction time by one step synthesis. [n(In):n(MA):n(Zn):n(S)=1:3:1:1]

图 3 不同的In:MA物质的量比下, 不同反应时间的样品吸收光谱图 (采用一步法制备样品, n(In):n(P):n(Zn):n(S)=1:6:1:1)
Figure 3 The evolution of absorption spectra of the samples synthesized at different In:MA molar ratios with reaction time by one step synthesis. [n(In):n(P):n(Zn):n(S)=1:6:1:1]

图 4 (A) 改变反应最初加入的In:Zn (S) 物质的量比, 合成的不同InPZnS核的吸收光谱图[n(In):n(P):n(MA)=1:6:3];采用两步法包覆, 不同反应时间的样品吸收光谱图 (B) 和荧光光谱图 (C)
Figure 4 (A) The absorption spectra of the samples synthesized at different In:Zn (S) molar ratios with reaction time. [n(In):n(P):n(MA)=1:6:3]. The evolution of absorption (B) and PL (C) spectra of the samples synthesized by two-step synthesis with reaction time

图 5 (A) 不同粒径的InPZnS/ZnS核壳量子点在紫外灯照射下的图像; (B) 不同粒径的InPZnS/ZnS量子点吸收和荧光光谱图
Figure 5 (A) The photograph of different sized InPZnS/ZnS QDs under UV light. (B) The PL (λex 450 nm) and absorption spectra of those different sized InPZnS/ZnS QDs

图 6 (A) InPZnS核的透射电镜图 (λem=650 nm); (B) 一步法合成的InPZnS/ZnS量子点的透射电镜图 (λem=577 nm, In:P:MA:Zn:S=1:6:3:1:1); (C) 两步法合成的InPZnS/ZnS量子点的透射电镜图 (λem=680 nm, 成核过程: In:P:MA:Zn:S=1:6:3:0.15:0.15, In=0.1 mmol; 包覆过程:再注入1 mmol的Zn (St)2和1 mmol DDT)
Figure 6 (A) TEM images of the as-prepared InPZnS QDs. (λem=650 nm). (B) TEM images of InPZnS/ZnS QDs by one step synthesis. (λem=577 nm, In:P:MA:Zn:S=1:6:3:1:1) (C) TEM images of InPZnS/ZnS QDs by two-step synthesis. (λem=680 nm, Core synthesis: In:P:MA:Zn:S=1:6:3:0.15:0.15, In=0.1 mmol, Shell synthesis: 1 mmol Zn (st)2, 1 mmol DDT and 5 mL ODE)

图 7 (A) InP纳米晶核的X射线粉末衍射光谱图 (XRD); (B) InPZnS纳米晶核的XRD图; (C) 一步法制备的InPZnS/ZnS核壳量子点的XRD图; (D) 采用两步法制备的InPZnS/ZnS核壳量子点的XRD图
Figure 7 (A) Powder X-ray Diffractograms of InP QDs. (B) Powder X-ray Diffractograms of InPZnS QDs. (C) Powder X-ray Diffractograms of InPZnS/ZnS QDs by one step synthesis. [n(In):n(Zn)=1:1] (D) Powder X-ray Diffractograms of InPZnS/ZnS QDs by two-step synthesis. [n(In):n(Zn)=1:1] For comparison the diffraction patterns of bulk cubic InP (triangles) and ZnS (squares) are displayed.

图 8 改变磷前体, 采用两步法合成的不同时间的样品吸收光谱图 (A) 和荧光光谱图 (B)
Figure 8 The evolution of (A) absorption spectra and (B) PL spectra of the samples with reaction time by two-step synthesis

图 9 合成InPZnS/ZnS核壳量子点的实验装置图
Figure 9 Experimental Setup for the Synthesis of InPZnS/ZnS QDs
表 1 不同反应时间的样品荧光峰的位置、量子产率和荧光峰半峰宽值
Table 1. The quantum yield (QY) and full-width at half-maximum (FWHM) of the as prepared samples with reaction time
Time 5 min 10 min 20 min 30 min 35 min 40 min 50 min 60 min 70 min 90 min Peak/nm 552 559 565 564 563 567 569 570 572 577 QYs/% 0.70 2.20 6.00 9.20 30.0 31.0 32.0 37.0 44.0 48.4 FWHM/nm 72.0 67.2 62.0 60.0 58.0 58.8 58.0 59.4 58.8 58.8  下载: 导出CSV
下载: 导出CSV
表 2 X射线能谱图 (EDX) 中不同包覆时间的InPZnS/ZnS量子点的各种元素的百分比: (A) 采用一步法, 各元素物质的量比为n(In):n(P):n(MA):n(Zn):n(S)=1:1:3:1:1时, 30, 40, 60, 90 min的样品; (B) 两步法制备的样品:成核过程, 各元素物质的量比为n(In):n(P):n(MA):n(Zn):n(S)=1:1:6:0.15:0.15; 30 min后, 补充加入一定量的锌和硫前体进行ZnS包覆. [n(In):n(Zn):n(S)=1:10:10]
Table 2. The ratio of these elements of samples with different shell thickness from the EDX spectra (JEOL JSM-840A SEM equipped with an Oxford Instruments energy dispersive X-ray analyzer): (A) synthesized by one step synthesis: n(In):n(P):n(MA):n(Zn):n(S)=1:1:3:1:1, 30, 40, 60, 90 min; (B) synthesized by two-step synthesis: Core synthesis, n(In):n(P):n(MA):n(Zn):n(S)=1:1:6:0.15:0.15, 30 min, and coated ZnS shell by adding plus Zinc and sulfur precursors. [n(In):n(Zn):n(S)=1:10:10]
Elt/A% A30 min A40 min A60 min A90 min B90 min P 41.16 40.48 33.05 32.42 8.67 S 10.77 12.18 16.89 22.35 47.39 Zn 10.19 12.92 17.74 16.68 35.87 In 37.88 34.42 32.32 28.55 8.07 Total 100.00 100.00 100.00 100.00 100.00
下载: 导出CSV
-










 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 31
- 文章访问数: 4010
- HTML全文浏览量: 859
 下载:
下载:
