引用本文:
张思伟, 张俊, 吴思达, 吕伟, 康飞宇, 杨全红. 钠离子电池用碳负极材料研究进展[J]. 化学学报,
2017, 75(2): 163-172.
doi:
10.6023/A16080428

Citation: Zhang Siwei, Zhang Jun, Wu Sida, Lv Wei, Kang Feiyu, Yang Quan-Hong. Research Advances of Carbon-based Anode Materials for Sodium-Ion Batteries[J]. Acta Chimica Sinica, 2017, 75(2): 163-172. doi: 10.6023/A16080428
Citation: Zhang Siwei, Zhang Jun, Wu Sida, Lv Wei, Kang Feiyu, Yang Quan-Hong. Research Advances of Carbon-based Anode Materials for Sodium-Ion Batteries[J]. Acta Chimica Sinica, 2017, 75(2): 163-172. doi: 10.6023/A16080428
钠离子电池用碳负极材料研究进展
摘要:
相较于目前主流的锂离子电池,钠离子电池成本相对较低,因而有望在未来大规模储能系统中获得重要应用,然而其实用化进程仍受制于缺少合适的正负极材料,特别是性能优异且实用化的负极材料.钠离子电池与锂离子电池具有相似的工作原理,但钠离子和锂离子在碳负极材料中的储存行为却有着很大的不同.总体而言,碳材料仍是目前最有望促进钠离子电池实用化的关键负极材料.本文系统总结并分析了目前已有碳材料中钠离子的储存机制,对负极材料的设计思路和研究进展进行了概述,着重阐述了商用化碳分子筛在钠离子电池中的实用化前景.最后,本文对钠离子电池中碳负极材料的未来发展方向进行了展望.
English
Research Advances of Carbon-based Anode Materials for Sodium-Ion Batteries
Abstract:
Compared with the widely-used lithium-ion battery (LIB), sodium-ion battery (SIB) is a promising energy storage device for large scale energy storage systems due to the low cost and environmental benignity of sodium. However, its practical use is restricted by the lack of suitable anode and cathode materials, especially the applicable anode materials with high performance. SIBs have similar working mechanism to LIBs, and thus, carbon materials are the most promising anode materials for SIBs. But the storage behaviors of Na+ and Li+ in carbon-based anodes are quite different. Graphite, which is used as the anode of commercial LIBs, hardly accommodates sodium ions. Thus, many researchers investigated sodium ion storage in disordered carbons, especially the hard carbons. Hard carbon is composed of disordered turbostratic nanodomains (TNs) and the pores formed between these domains. The edge/defect sites on the carbon surface, e.g., carbenes, vacancies, and dangling bonds on the edges of TNs, the interlayer space in TNs, and the pores can host the sodium ions. High porosity is normally needed to reach a high capacity and rate capability. But this leads to large irreversible reactions, and thus, a low initial Coulombic efficiency and poor cyclic stability. In this paper, sodium ion storage behaviors in different carbon structures are discussed and the design principles and research advances of carbon-based anode materials are reviewed. Particularly, the commercial carbon molecular sieve (CMS) is highlighted as a promising anode material for the practical use of SIBs. Finally, the future development of carbon anodes for SIB is commented and prospected.
-
Key words:
- carbon materials
- / sodium ion battery
- / storage mechanism
- / carbon molecular sieves
-
1 引言
随着电动车产业以及可再生能源(如风能、太阳能等)在世界范围内的快速发展, 大规模的能源储存技术已经成为制约其可持续发展的关键, 也是未来解决风能及太阳能等可再生能源不连续性与能源需求连续性矛盾的主要途径. 以锂离子电池为代表的电化学储能系统因能量密度和效率高等优势, 成为储能领域的宠儿[1~3]. 然而锂的储量有限且分布不均使锂离子电池成本较高, 而且频繁充放电使其寿命缩减, 导致在基站式储能等大规模储能领域中投资成本居高不下, 难以满足未来规模化应用需求[4, 5]. 与锂同族的碱金属元素钠, 具有与锂相似的性质(如表 1所示), 并且储量丰富, 成本较低. 另外, 钠与铝之间不发生合金化作用, 故可取代铜箔做钠离子电池负极的集流体, 进一步降低了电池成本, 因此以钠离子为电荷载体的钠离子电池在大规模储能系统中具有更大的应用潜力[6], 吸引了国内外研究者广泛的关注[7, 8].
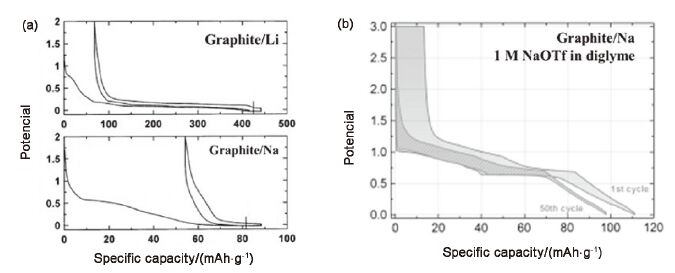
在二十世纪七、八十年代, 钠离子电池与锂离子电池的研究几乎处于同一水平, 然而石墨负极在锂离子电池中的成功应用直接促进了其商业化进程, 钠离子电池却至今仍未实现产业化突破, 瓶颈之一是缺少合适的实用化负极材料[10]. 近几年钠离子电池的研究相继取得重要的进展, 其中负极材料的研究主要集中于碳材料以及一些非碳材料(金属及氧化物材料、合金材料及磷等)[11]. 非碳材料对锂和钠都表现出高的存储容量, 但是由于导电率低、体积变化大和易粉化等问题, 即便在商业化程度很高的锂离子电池中仍未获得大规模应用. 上述问题在钠离子电池中同样存在, 而碳基材料不仅具有较低的嵌钠平台, 较高的容量和良好的循环稳定性, 还具有资源丰富, 制备简单等优点. 因此碳材料仍是最有希望推动钠离子产业化的关键负极材料[12~14].
目前具有应用前景的主要储钠碳材料包括: (1)软碳材料, 主要指具有层状结构的类石墨碳材料, 主要以层间插层的形式储钠, 导电性较好, 但是比容量较低; (2)硬碳材料, 结构复杂多样, 通常具有多种储存钠离子的形式, 虽然容量较高, 但是首次库仑效率低; (3)纳米碳材料, 诸如石墨烯、碳纳米管等, 通常主要依靠表面吸附实现储钠, 可以实现快速充放电, 但是库仑效率低、循环性差等问题使其在短时间内也难以获得实际应用. 本文从已有的不同储钠机理出发, 分析了不同碳材料的储钠过程, 归纳和总结了不同碳材料的结构设计和研究进展.
2 钠离子在碳负极材料中的储存机理
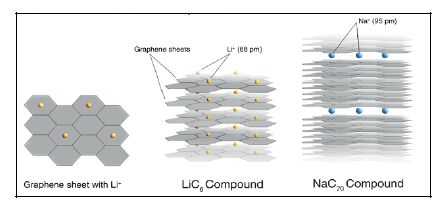
锂离子在负极碳材料中的存储主要有4种机制: (1)插层嵌入机制: 锂离子插入到石墨烯片层面之间, 形成锂-石墨层间化合物[15~17](如图 1a), 且石墨烯平面尺寸愈大(La大), 插入的锂量愈多; (2)分子储锂机制: 也称过剩储锂行为, 即在石墨化程度较低的碳片层间形成Li2分子(如图 1b); (3)微孔储锂机制: 在富含孔隙的碳结构中, 以微孔填充的形式达到储锂的目的; (4)“卡片屋”机制: 即由碳片层乱堆形成的结构中, 兼有上述多种方式的储锂过程, 包括乱堆片层中插入锂、片层边缘储锂和碳层表面吸附储锂等方式[18~22].
钠离子与锂离子具有相似的物理化学性质和储存机制, 但对于不同的碳材料, 钠离子存储和嵌入行为又表现出不同于锂离子的特点[24]. 目前报道的主要三种钠离子储存机制包括插层嵌入、微孔填充和“卡片屋”模型机制. 本节将重点介绍上述几种储钠机制.
2.1 插层嵌入机制
钠与锂在类石墨材料的层间储存行为不同. 如图 2所示, 锂离子嵌入石墨类负极后形成稳定的一阶插层化合物LiC6, 理论容量为372 mAh·g-1. 由于钠离子的半径较锂离子大, 且由于热力学原因无法与石墨形成一阶稳定的插层化合物(形成NaC70), 因此石墨的储钠活性较低, 理论容量仅为31 mAh·g-1[25]. 理论计算表明可实现钠离子插入的最小石墨层间距为0.37 nm[26]. 因此, 克服片层之间的范德华力, 增大石墨类碳材料的层间距, 使钠离子嵌入其中是类石墨层状材料实现钠离子存储的主要设计思路, 目前思路已得到实验验证并取得重要的研究进展[27~31]. Wang等[28]报道了部分还原的氧化石墨制备的膨胀石墨作为钠离子电池的负极(如图 3所示), 其层间距为0.43 nm, 在20 mA·g-1的电流密度下比容量约300 mAh·g-1, 并且在100 mA·g-1的高电流密度下依然具有良好的循环性能. 通过对比不同的还原条件, Singh等[27]证明增大层间距确实有利于提高层间储钠能力. 除此之外, 可石墨化的软碳在经过适当的热处理后, 也可以获得较大的层间储钠容量. Mitlin等[30]以泥煤苔为前躯体制备出石墨化碳, 其层间距为0.388 nm, 表现出优异的储钠能力; Ji等[31]也发现可石墨化软碳的储钠能力随层间距的增大而增大. 但是膨胀石墨及热处理后软碳的充放电曲线是一个平缓的斜坡, 并没有明显的脱嵌钠平台, 这与锂离子在石墨类材料中的储存行为极不相同.
 图 3
(a~c)钠离子在膨胀石墨中的储存示意图, (d)循环性能和库仑效率, (e)充放电曲线(其中PG指原始石墨, GO指氧化石墨, EG指膨胀石墨)[28]
Figure 3.
(a~c) Illustration of Na+ion storage in graphite-based materials; (d) Cycle performance and coulombic efficiency of the sodium cell; (e) Charge/discharge profiles of pristine graphite (PG), graphite oxides (GO), expanded graphite (EG)[28]
图 3
(a~c)钠离子在膨胀石墨中的储存示意图, (d)循环性能和库仑效率, (e)充放电曲线(其中PG指原始石墨, GO指氧化石墨, EG指膨胀石墨)[28]
Figure 3.
(a~c) Illustration of Na+ion storage in graphite-based materials; (d) Cycle performance and coulombic efficiency of the sodium cell; (e) Charge/discharge profiles of pristine graphite (PG), graphite oxides (GO), expanded graphite (EG)[28]
2.2 “卡片屋”模型机制
2000年, Stevens和Dahn首次以葡萄糖为前躯体制备的硬碳材料应用到钠离子电池中[23], 获得了约300 mAh·g-1的比容量. 研究者们据此提出了“卡片屋”模型的储钠机制(如图 4), 这种模型与锂离子存储中的“卡片屋”模型基本一致[24]. 如图 4所示, 在1.0~0.2 V的斜坡容量主要是钠离子嵌入到石墨烯片形成的层间贡献的, 这与膨胀石墨的充放电曲线类似, 嵌入电位随着嵌入量的增加而降低; 而小于0.2 V(接近钠金属的沉积电位)的平台容量是由于乱层堆垛的纳米级石墨微晶(Turbostratic nanodomains)形成的孔洞吸附和钠金属沉积贡献的.
受Stevens和Dahn工作的启发, 众多的研究者将目光聚焦在以“卡片屋”模型为储钠机制的硬碳材料上, 并取得了一系列成果[32~35]. Komaba等[32]使用XRD发现在0.2 V时, 硬碳的(002)峰从23.4°移动到21.0°, 表明层间距增大, 可能是钠离子嵌入平行排列的石墨层间导致的, 而再次充电至2.0 V时, 峰位又回到初始位置, 说明钠离子可以从石墨层间可逆的脱嵌, 他们利用SAXS研究了硬碳材料在不同电位下微孔内部的电子密度的变化, 在小于0.2 V时, SAXS检测到0.03~0.07 Å-1范围内微孔电子密度明显下降, 即低电位的容量对应于钠离子在微孔中的脱嵌. 同时Raman也表现出相似的结果, 在斜坡区域内, 硬碳材料的G峰出现明显的右移, 而在低电压平台区域, G峰并没有移动, 表明斜坡容量对应的是钠离子嵌入碳层的过程. 相似的储钠行为也在一维(1D)碳材料中得到体现[36, 37].
虽然部分结果支持“卡片屋”模型机理, 但一些实验现象与此机理存在明显的冲突. 如低温热解硬碳存在大量的微孔, 但几乎没有低电压平台, 而随着热解温度升高, 硬碳材料的微孔容量减小, 但是平台容量却增加. Cao等在2012年首次提出低电位对应钠离子在石墨层间的脱嵌行为, 而高电压斜坡区域对应钠离子在硬碳表面的吸附行为. 他们发现聚苯胺热解管状碳的储钠和石墨储锂的充放电曲线非常相似, 而该材料的储钠和储锂行为差异很大, 由此推测聚苯胺热解碳管在0~0.2 V低电压平台的储钠机理与石墨储锂应该类似, 是碱金属离子在石墨层间的脱嵌形成嵌入化合物的过程. 他们通过理论计算验证了此想法, 发现适合钠离子嵌入的石墨层间距约为0.37 nm[26]; Mitlin等使用原位XRD测试分别证实了乱层堆垛的石墨微晶在低电压下层间的膨胀行为[38~41]. 在此基础上, Ji等[42]提出了新的“卡片屋”储钠模型(如图 5): 在1.0~0.2 V的斜坡容量是由钠离子在乱层堆垛的石墨微晶边缘和缺陷的化学吸附; 在较低的平台0.2~0.01 V是由钠离子嵌入到石墨微晶层间引起的; 而小于0.01 V的容量是由于钠离子在石墨微晶形成的孔洞的吸附作用引起的.
随后, Mitlin等也证明了这一机理. 他们用天然泥煤苔为前躯体, 在不同温度下热解, 实验发现随着热解温度升高, 石墨烯片层之间的层间距逐渐减小, 同时石墨微晶的长度La、宽度Lc均明显增加, 拉曼表征的G峰和D峰的比值也增大. 在HRTEM下观察到石墨微晶的长度明显变大, 并且排布趋于平整. 通过氮气吸脱附表征发现随着热解温度升高, 微孔逐渐减少. 结合电化学性质, 热解温度越高, 低电压平台容量越高, 这与石墨化程度成正比, 与微孔含量成反比, 暗示低电压平台对应钠离子在石墨烯层间的嵌入, 而不是微孔的吸附. 作为对比, 他们制备了含有丰富微孔的活性炭, 发现虽然微孔丰富, 但是依然不存在低电压区平台.
两种“卡片屋”模型对硬碳材料中钠离子存储的平台容量和斜坡容量的解释截然相反, 却又各自符合其所讨论的材料电化学特性与结构之间的对应关系. 结合锂离子电池的“卡片屋”模型及钠离子相关研究进展, 表面钠与锂在上述硬碳材料中的存储行为是有区别的, 钠离子具有自己的特殊性, 并且由于碳材料前躯体、结构、形貌的不同, 储钠行为也各不相同. 充分认识和理解硬碳材料的储钠机制, 仍需要对不同处理条件下硬碳材料的结构和电化学性能深入研究.
2.3 微孔储钠机制
微孔填充和介孔中的吸附是两种不同的吸附过程, 对应着不同的离子存储机制. Huang等总结了具有不同孔径的碳材料在双电层电容器中储能模型——当孔径小于2 nm时, 比容量的计算公式为
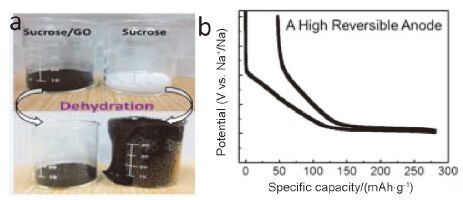
$C/A\frac{{{\varepsilon }_{r}}{{\varepsilon }_{0}}}{b\ln (b/{{a}_{0}})}$ ; 当孔径为2~50 nm时,$C/A\frac{{{\varepsilon }_{r}}{{\varepsilon }_{0}}}{b\ln (\frac{b}{b-d})}$ ; 当孔径大于50 nm时,$C/A\frac{{{\varepsilon }_{r}}{{\varepsilon }_{0}}}{d}$ . (其中εr:电解质介电常数, ε0:真空介电常数, A: 电极比表面积, d: 双电层的有效厚度(德拜长度), a和b分别为模型孔的内径和外径). 通过结合密度泛函理论计算活性碳、模板碳等多种碳材料与不同的电解液(有机电解液、无机电解液)之间的相互作用, 进一步证明这个模型的普适性[43]. 在钠离子电池中, 具有丰富微孔或中孔的碳材料, 由于较大的比表面积, 首次充放电过程的库仑效率普遍较低, 因此难以实用化. 微孔储钠要求碳材料孔径分布必须合理而且集中, 孔径太大将使得电解液分子与电极材料接触面积太大, 进而副反应较多, 造成首圈大量不可逆容量, 孔径过小则可能使钠离子无法进入和快速扩散.Ji等[44]以蔗糖为前驱体, 通过调控CO2活化的温度与时间, 获得孔隙率不同的碳材料, 研究发现通过降低孔容和比表面积可以有效提高可逆容量和库仑效率. 这可能是由于低的比表面积有利于在首次放电过程中在其表面形成一层完整的SEI膜, 减少了表面不可逆反应的发生. Tarascon等[45]以纳米碳纤维(CNFs)为研究对象, 证明在较高的温度(2000 ℃)热处理后, CNFs具有更优的储钠性能, 其脱嵌钠的平台约在0.1 V, 容量可达200 mAh·g-1, 这主要是由于该温度下CNFs较高的石墨化程度、低的活性比表面积以及部分介孔的存在.
最近我们报道了一种商业化碳材料——广泛用于氧/氮变压吸附分离的碳分子筛(CMS), 可以作为钠离子电池负极材料, 并表现出很好的储钠能力. 如图 6所示, 碳分子筛中的孔主要在极微孔范畴, 孔隙大小处于氮气可吸附孔径的临界值(约0.4 nm), 故使用不同的吸附质测试得到的比表面和孔径分布结果差距很大. 如图 7所示, 当吸附剂为氮气时, 吸脱附曲线为典型的Ⅱ型曲线, 表现出无孔的吸附行为, 比表面积仅为17 m2·g-1; 当吸附剂为二氧化碳时, 吸附曲线为典型的Ⅰ型曲线, 为典型的微孔吸附行为, 比表面积则可达到417 m2·g-1[46]. 小的孔径只允许钠离子进入而阻止了电解液分子进入, 从而降低了电解液和活性物质的接触面积, 减少了SEI膜的形成及其他副反应的发生, 提高了首次库仑效率, 如图 7所示.
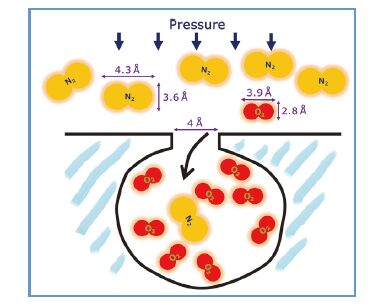 图 6
碳分子筛用于工业制氮的示意图
Figure 6.
Illustration of CMS for the produce of N2
图 6
碳分子筛用于工业制氮的示意图
Figure 6.
Illustration of CMS for the produce of N2
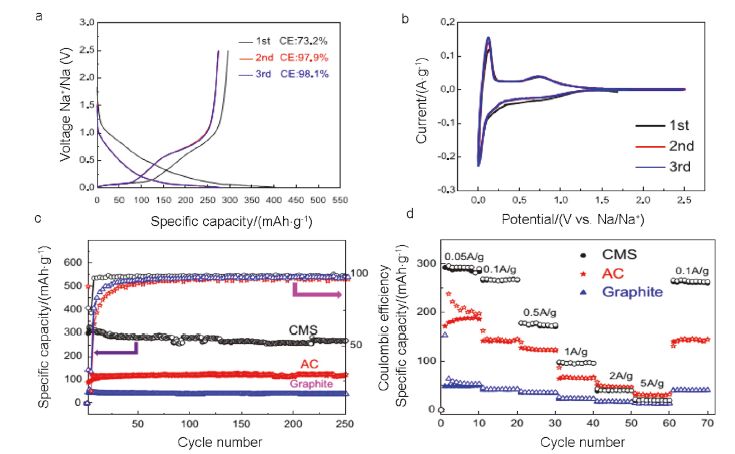 图 7
碳分子筛做负极材料的钠离子电池电化学性能: (a)前3圈充放电曲线; (b)前3圈的CV曲线; (c)电流100 mA·g-1时的循环性能; (d)不同电流下的倍率性能[46]
Figure 7.
Properties of SIB with the carbon molecule sieve (CMS) anode: (a) Charge/discharge curve of the first three cycles; (b) CV curve of the first three cycles; (c) Cycle performance at the current of 100 mA·g-1; (d) Rate capability at different current densities[46]
图 7
碳分子筛做负极材料的钠离子电池电化学性能: (a)前3圈充放电曲线; (b)前3圈的CV曲线; (c)电流100 mA·g-1时的循环性能; (d)不同电流下的倍率性能[46]
Figure 7.
Properties of SIB with the carbon molecule sieve (CMS) anode: (a) Charge/discharge curve of the first three cycles; (b) CV curve of the first three cycles; (c) Cycle performance at the current of 100 mA·g-1; (d) Rate capability at different current densities[46]
3 几种典型碳材料的设计思路与研究进展
如前所述, 不同种类碳材料储存钠离子的机理各不相同, 而这与碳材料的结构息息相关. 本节从上述三种储钠机理出发, 概述了目前碳材料的研究进展, 讨论了材料的结构对电池性能的影响, 并进一步提出了不同材料后期的结构设计和优化方向.
3.1 类石墨层状碳材料研究进展
在上一小结中我们提到, 对于石墨而言, 由于无法与钠离子形成稳定插层化合物, 因此不适于作为钠离子电池的负极材料. 通过扩大层间距的方法, 可以实现钠离子的插层, 但是这种情况下得到的比容量往往较低, 而且扩大层间距的方法也较为复杂. 近期, 另外一种可将石墨直接应用到钠离子电池负极中的方法得到了广泛的关注, 但这种方法并不是从电极材料入手而是从电解液角度. Adelhelm等[47]报道了一种非常规的钠离子-电解液共插层储钠机制. 不同于酯类电解液去溶解化的插层过程, 在醚基电解液中, 电解液中的醚类分子与钠离子可以同时插入石墨层间, 并形成稳定的插层化合物Na(diglyme)2C20, 有效的提高了钠离子在石墨中的嵌入量, 在37 mA·g-1的电流密度下可逆容量达到约100 mAh·g-1, 并在0.6 V附近具有明显的脱嵌钠平台, 这与锂离子在石墨中的嵌入插层反应平台(约0.1 V)不同(如图 8a). 值得注意的是, 基于这种共插层机制的钠离子电池首圈库仑效率接近90%, 是目前报道的最高值. 这一研究打破了石墨不能应用于钠离子电池的传统观念, 但其容量仍然有待提升, 较高的充放电平台也限制了它的实际的应用. Kang等[48]进一步研究了天然石墨在不同种类的醚基电解液中的插层过程, 研究发现钠离子-电解液共插层的现象伴随着赝电容行为, 可以进一步提升比容量, 而且不同的种类的电解液对电池的倍率性能及负极材料的储钠过程有极大的影响. 在同一种溶剂中使用不同的钠盐(NaPF6、NaClO4、NaCF3SO3)对石墨的储钠行为并没有明显的影响. Pint等[49]进一步将醚基电解液应用到层数在2~9层的石墨烯中, 证明钠离子与电解液共插层的储钠形式省去了去溶剂化的过程, 具有更快的储钠能力, 从而具有更好的倍率性能. 如图 9所示, 在30 A·g-1的高电流密度下, 该石墨烯材料依然保持了约100 mAh·g-1的比容量, 在0.6~0.7 V之间存在明显的充放电平台. 使用与钠离子可实现共插层的电解液, 提高首次库仑效率与容量是未来钠离子电池的重要研究方向之一. 但是钠离子-溶剂共插层所需电位较高(通常为0.6 V左右), 插层过程中产生近300%的体积膨胀[50], 不利于实际应用.
 图 9
少数片层的石墨烯在醚基电解液中的储钠行为: (a)前5圈充放电曲线; (b)不同倍率充放电曲线对比; (c)倍率性能; (d)循环性能; (e)8000圈循环后的充放电曲线[49]
Figure 9.
Sodium storage behavior of few-layer graphene (FLG) electrode with diglyme electrolyte: (a) First five charge/discharge profiles at galvanostatic current density of 0.2 A·g-1. (b) Charge/discharge profiles at galvanostatic current densities ranging from 1 A·g-1to 30 A·g-1 and (c) the corresponding cycling performance, inset shows the linear relation between specific capacity and current density. (d) Extended cycling performance at current density of 12 A·g-1over 8000 cycles with (e) a selected galvanostatic charge/discharge profiles, inset shows that the over potential decreases with cycling[49]
图 9
少数片层的石墨烯在醚基电解液中的储钠行为: (a)前5圈充放电曲线; (b)不同倍率充放电曲线对比; (c)倍率性能; (d)循环性能; (e)8000圈循环后的充放电曲线[49]
Figure 9.
Sodium storage behavior of few-layer graphene (FLG) electrode with diglyme electrolyte: (a) First five charge/discharge profiles at galvanostatic current density of 0.2 A·g-1. (b) Charge/discharge profiles at galvanostatic current densities ranging from 1 A·g-1to 30 A·g-1 and (c) the corresponding cycling performance, inset shows the linear relation between specific capacity and current density. (d) Extended cycling performance at current density of 12 A·g-1over 8000 cycles with (e) a selected galvanostatic charge/discharge profiles, inset shows that the over potential decreases with cycling[49]
值得深思的是, 锂离子-电解液共插层是限制醚基电解液在锂离子电池中应用的主要原因——这种共插层造成石墨片层在长循环中的剥离, 导致锂离子电池的循环稳定性差[51~53], 该现象并没有在钠离子电池中发现. 实际上, 早在1950年, 科学家就发现碱金属、醚、石墨可以相互作用形成稳定的Na(NH3)2C12, 在目前钠离子电池研究中形成的化合物主要是Na(diglyme)2C20, 从这个意义上说, 石墨应用于钠离子电池负极材料的容量还有可能得到提升. 相关研究也启示钠离子电池的研究者: 也许在锂离子电池上应用失败的材料在钠离子电池上会有很好的应用前景.
3.2 硬碳材料结构设计与研究进展
以“卡片屋”模型储存钠离子的碳材料主要是硬碳. 相比于具有层状结构的碳材料, 由石墨微晶乱层堆垛形成的硬碳具有发达的纳米孔隙和一次颗粒间的空隙, 增加了钠离子储存活性位, 并且相比于插层储钠过程, 孔隙吸附作用可以提高碳材料的倍率性能. 增大孔容积是提高硬碳材料性能的一个主要研究方向. Wenzel等[54]通过以二氧化硅为模板和中间相沥青为碳源合成了具有层次孔结构的碳材料, 缩短了钠离子的传输路径, 在5C的高倍率下可获得100 mAh·g-1的可逆容量(如图 10). 但是提高孔容积会引起一个不可忽视的缺点——极低的首次库仑效率. 大多数碳负极材料在钠离子电池中的首次库仑效率只有15%~45%. 和锂离子电池相似, 大的不可逆容量主要是由于负极材料高的比表面积导致电解液在负极材料表面还原形成较多的SEI膜以及不可逆反应. 因此硬碳材料设计中必须要降低电极材料与电解液的接触面积, 这一思路也已经取得重要进展[55~57]. Ji等[57]在蔗糖焦化反应过程中加入氧化石墨烯(graphene oxide, GO)抑制蔗糖发泡(如图 11所示), 经过1000 ℃的高温热处理, 得到低比表面积的碳材料; 随着热处理温度从1000 ℃升至1600 ℃, 材料的比表面积由137 m2·g-1降至5 m2·g-1, 相应的, 首循环库仑效率由74%提升至83%.
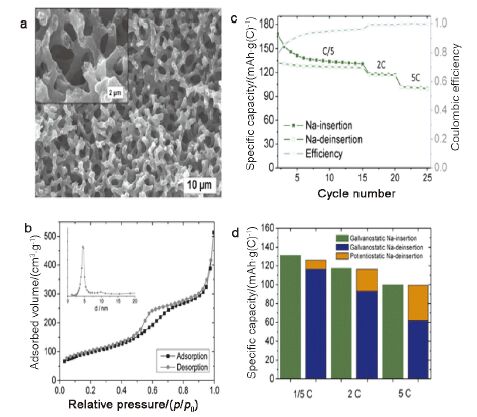 图 10
(a)互相连通的大孔结构的SEM图; (b)样品吸脱附曲线与孔径分布; (c)倍率性能; (d) 0~1.6 V电压区间的比容量[51]
Figure 10.
(a) SEM image of the inter connected macroporous structure. (b) Adsorption isotherm and pore size distribution derived from nitrogen adsorption. (c) Rate capability. (d) Contribution of the potentiostatic step at 1.6 V to the total charge capacity upon Na-extraction[51]
图 10
(a)互相连通的大孔结构的SEM图; (b)样品吸脱附曲线与孔径分布; (c)倍率性能; (d) 0~1.6 V电压区间的比容量[51]
Figure 10.
(a) SEM image of the inter connected macroporous structure. (b) Adsorption isotherm and pore size distribution derived from nitrogen adsorption. (c) Rate capability. (d) Contribution of the potentiostatic step at 1.6 V to the total charge capacity upon Na-extraction[51]
Hu等[58]发现在热处理前用2, 2, 6, 6-tetramethylpi- peridine-1-oxyl (TEMPO)预处理纤维素, 可将碳化获得的材料比表面积由586 m2·g-1降至126 m2·g-1, 首次库仑效率也由28%提升至72%, 并且在100 mA·g-1的电流密度下获得200 mAh·g-1的可逆容量. 上述低比表面积的碳负极材料通常具有更宽的电化学窗口, 具有较高的实用价值. 随后其课题组又利用沥青和酚醛树脂以不同比例在不同温度下热解, 得到极低比表面积、化学性能优异的负极材料. 不仅库仑效率高达82%, 并且可逆容量可达254 mAh·g-1. 沥青与酚醛树脂原料便宜易获取, 并且收率高, 为商业化大生产提供了可能. 其课题组也尝试使用无烟煤热解碳[56]、天然棉花[59]热解产物作为钠离子电池负极材料, 均得到良好的数据.
值得思考的是, 低比表面积并不意味着一定可以获得较大的首次库仑效率. 早在1999年, Billaud等以树脂为前躯体制备了一种比表面积仅为1 m2·g-1的碳纤维负极材料, 其首次的库仑效率仅为30%. Billaud等[60]认为这种低的库仑效率是由于大量的钠离子“不可逆陷入”到相邻石墨微晶之间的纳米孔洞中无法“逃脱”造成的. 在钠离子电池中, 很多种碳材料首次库仑效率都较低, 但是第二圈库仑效率即能接近100%, 因此造成首次不可逆容量的具体原因仍然需要进一步的实验与理论探索.
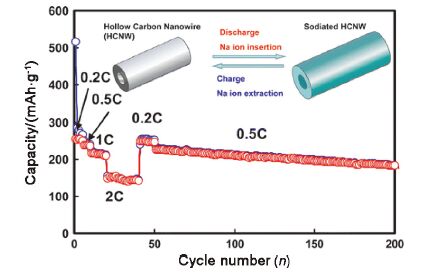
碳材料的微观形貌和结构也在电化学储钠中有重要的作用, 包括石墨烯在内众多的纳米碳已经应用到钠离子电池[61~68]中并表现出与锂离子电池相似的优势: 较好的倍率性能和较高的容量[69]. 由于纳米线、纳米管和纳米片等结构具有较大的比表面积, 能有效减小离子的扩散路径, 因此纳米结构设计能有效改善碳基材料的电化学性能. 与之前报道的碳基材料相比, 具有一维或者二维纳米结构的碳材料拥有更优异的储钠性能. 碳纳米管(CNTs)、碳纳米纤维(CNFs)、碳衍生物(CDCs)等纳米结构在钠离子电池体系中也受到了重视. Cao等[26]运用自组装法合成了聚苯胺中空碳纳米线前驱体, 然后在高温下碳化得到中空碳纳米线(如图 12所示). 在50 mA·g -1的电流密度下, 经过400次循环后仍保持206 mAh·g-1.
Ji等[42]也报道了碳片层、微孔及其边缘、表面缺陷(如空位、碳层边缘、自由键和碳烯等)三种储钠位点. 他们在1100、1400和1600℃下将蔗糖热解, 并将产物与玻璃碳(热解温度超过2000 ℃)进行对比. 通过XRD与Raman表征发现石墨微晶的长度La随热解温度升高逐渐变大, 基本满足线性回归(R2=0.95), ID/IG逐渐减小, 而随着温度升高, 斜坡容量逐渐减少, 与ID/IG比值变化一致, 由此可以说明斜坡容量是由硬碳材料的缺陷贡献的; 为了进一步证明该说法, 他们还利用了恒电流间歇滴定技术(GITT)测试了钠离子电池在不同电压范围内的扩散系数, 发现钠离子扩散系数在斜坡区域大于平台区域, 而石墨层边缘及表面缺陷相比于石墨层间嵌入更容易发生, 即低电位平台对应钠离子在碳片层间的嵌脱, 斜坡区容量对应碳边缘或表面缺陷的储钠行为.
此外, 通过N、S、P等元素的掺杂可以改变碳的微观结构和电子状态, 进而影响碳材料的导电性和缺陷数量, 可进一步提升其储钠性能[69]. 但是上述纳米材料大的比表面积往往导致首圈库仑效率低, 而且能量密度低, 难以实用化. 我们小组通过毛细蒸发制备出高体积能量密度的石墨烯基材料应用于钠离子电容器中, 为纳米材料在电池中的应用提供了较为理想的解决方案[70].
除人工合成硬碳的前躯体外, 自然界中许多天然的有机物也是制备硬碳材料的良好前躯体, 这些天然有机物来源广泛、价格低廉, 环境友好. Li等[71]利用树叶的天然结构制备了无需粘结剂和集流体的极片, 在10 mA·g-1的电流密度下, 首圈库仑效率达到75%, 容量为360 mAh·g-1, 并且具有较好的倍率性能. Lotfabad等[40]利用香蕉皮热解后制备的硬碳材料在50 mA·g-1的电流密度下, 获得的可逆容量约为221 mAh·g-1, 且循环600圈后仍能保持93%. 该研究团队利用花生壳[41]和泥煤苔[30]作为前驱体制备的钠离子负极材料均表现出优异的循环性能. Jin等[72]热利用木质素作为碳源制备的碳纳米纤维网可逆容量可达292 mAh·g-1, 且在800 mA· g-1的电流密度下可逆容量达到130 mAh·g-1, 表现出优异的倍率性能. 此外, 苹果皮[73]、棉花[58]、木质素[74]等天然有机物热解后也均表现出较高的容量.
3.3 分子筛类碳材料结构设计与研究进展
如前所述, 依靠微孔填充存储钠离子的过程中, 要尽量避免溶剂分子的进入, 从而防止不可逆反应的发生以及库仑效率的降低. 我们研究团队[46]报道的碳分子筛恰好是符合上述要求一种碳材料. 将未经修饰改性的碳分子筛应用到钠离子电池负极中便表现出优异的储钠性能: 首次库仑效率73.2%, 在100 mA·g-1的电流密度下比容量约为280 mAh·g-1, 是目前报道过的商业化材料中储钠能力最优的, 甚至优于实验室中大多数经过复杂条件合成的硬碳材料. 如图 13所示, 使用商业化碳分子筛作为钠离子电池的负极的首次库仑效率高达73.2%, 而使用孔径稍大的商业活性碳材料, 首次效率只有22.1%, 同样条件下石墨的首次效率也只有32.0%. 碳分子筛中只存在大量的极微孔(孔径在0.4 nm左右), 只允许去溶剂化的钠离子进入, 有效减小了电解液和碳表面的接触面积, 减少了SEI膜的形成及其他副反应的发生, 提高了首次库仑效率. 商业用活性碳也具有丰富的孔隙, 但是其孔径较大(孔径在1 nm左右), 因此虽然首次放电容量高, 但是库仑效率极低(20%左右).
从钠离子电池商业化的角度考虑, 碳分子筛不仅是一种已大规模商用化的碳材料, 而且依靠微孔储钠不仅容量高, 并且在0.1 V附近具有较长的平台, 相比于“卡片屋”模型与插层储钠具有更大的实际应用潜力. 但是这种极微孔在电化学储能过程中必须对孔道的连通性改善, 缩短离子的传输路径, 进一步提高倍率性能. 因此, 对碳分子筛进行改性, 进一步提高其首圈的库仑效率并提升其容量及倍率性能, 将有望促进钠离子电池的真正商用化.
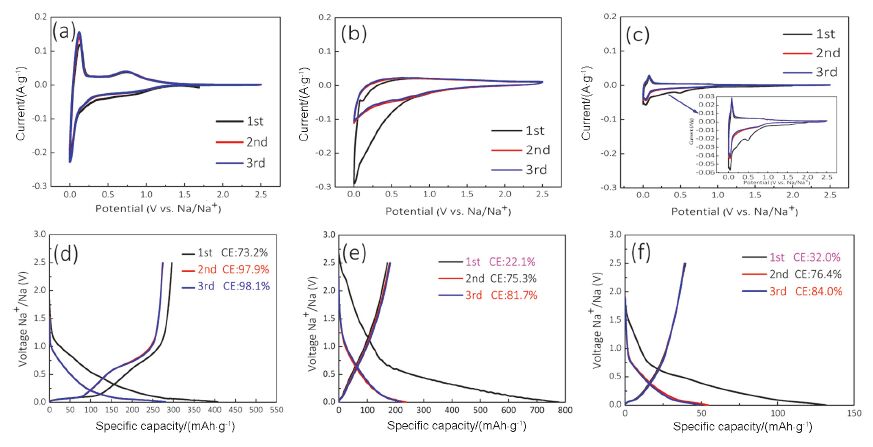 图 13
碳分子筛与石墨、活性碳(AC)在钠离子电池中应用的电化学性质对比: (a, d)碳分子筛的CV曲线与前三圈充放电曲线; (b, e)活性碳的CV曲线与前三圈充放电曲线; (c, f)石墨的CV曲线与前三圈充放电曲线[46]
Figure 13.
Electrochemical behaviors of CMS and the reference samples. (a, d): CMS; (b, e): AC; (c, f): Graphite. (a~c) CV curves at scan rates of 0.1 mV·s-1 and (d~f) the charge-discharge profiles for the first three cycles at a current density of 100 mA·g-1[46]
图 13
碳分子筛与石墨、活性碳(AC)在钠离子电池中应用的电化学性质对比: (a, d)碳分子筛的CV曲线与前三圈充放电曲线; (b, e)活性碳的CV曲线与前三圈充放电曲线; (c, f)石墨的CV曲线与前三圈充放电曲线[46]
Figure 13.
Electrochemical behaviors of CMS and the reference samples. (a, d): CMS; (b, e): AC; (c, f): Graphite. (a~c) CV curves at scan rates of 0.1 mV·s-1 and (d~f) the charge-discharge profiles for the first three cycles at a current density of 100 mA·g-1[46]
4 结论
随着新能源产业的快速发展, 储能市场也将逐步打开, 钠离子电池将有可能成为未来储能市场一颗闪耀的明星. 碳材料作为钠离子电池负极材料的研究已经取得了诸多成果, 但也面临着诸多问题. 如由于层间距不匹配, 石墨类碳基材料不适宜作为高性能钠离子电池负极, 而石墨化程度较低的碳材料具有相对较高的容量, 但循环稳定性能不佳. 材料的微观结构和钠离子脱嵌过程中的材料结构变化直接影响着钠离子电池的电化学性能, 但是目前相关研究仅停留在简单的体系甄选, 对钠离子脱嵌机制尚需要进一步研究, 只有深入的机理研究才能有效指导新材料的开发和性能的改进. 从商用化角度来看, 碳分子筛作为一种新型的钠离子电池负极材料兼具了高容量、高库仑效率以及长循环寿命等优点, 同时它也是一种已经得到大规模制备和商业应用的碳材料, 与石墨直接促进锂离子电池实用化的过程相类似, 碳分子筛也将有望在较短的时间内促进钠离子电池的实用化. 虽然如此, 碳分子筛的储钠机制也还未明确, 仍需深入的研究.
此外, 在后续新型碳基负极材料开发过程中, 应从以下几个方面展开: (1)从分子水平控制材料的微观结构, 设计更适合钠离子快速脱嵌的结构, 从根本上提高Na+的储存容量和脱嵌速率; (2)从去溶剂化以及SEI膜形成过程出发, 深入理解负极材料表面的电化学反应, 并针对不可逆反应提出抑制方法, 提高库仑效率以及循环稳定性; (3)提高电解液的离子电导率, 同时优化组分改善其与负极嵌钠材料的相容性和稳定性. 随着研究的不断深入, 上述关键问题将逐步得到解决或者显著改善, 才可真正的推动钠离子电池的商用化.
-
-
[1]
Tarascon, J.-M.; Armand, M. Nature 2001, 414(6861), 359.
-
[2]
Wu, X.; Jiang, L.; Cao, F.; Guo, Y.; Wan, L. Adv. Mater. 2009, 21(2710), 25.
-
[3]
Jung, H.; Jang, M.-W.; Hassoun, J.; Sun, Y.; Scrosati, B. Nat. Commun. 2011, 2(516), 638.
-
[4]
Goodenough, J.-B. Energy Storage Mater. 2015, 1, 158. doi: 10.1016/j.ensm.2015.07.001
-
[5]
Wang, Y.; Chen, R.; Chen, T.; Lv, H.; Zhu, G.; Ma, L.; Wang, C.; Jin, Z.; Liu, J. Energy Storage Mater. 2016, 4, 103.
-
[6]
Kubota, K.; Komaba, S. J. Electrochem. Soc. 2015, 14(162), A2538.
-
[7]
Xiang, X.; Lu, Y.; Chen, J. Acta Chim. Sinica 2012, 70, 32(in Chinese). (向兴德, 卢艳莹, 陈军, 化学学报, 2012, 70, 32.)
-
[8]
Li, H.; Wu, C.; Wu, F.; Bai, Y. Acta Chim. Sinica 2014, 72, 21(in Chinese). (李慧, 吴川, 吴锋, 白莹, 化学学报, 2014, 72, 21.)
-
[9]
Pan, H.; Hu, Y.; Chen, L. Energy Environ. Sci. 2013, 6(8), 2338.
-
[10]
Yabuuchi, N.; Kubota, K.; Dahbi, M.; Komaba, S. Chem. Rev. 2014, 114(23), 11636.
-
[11]
Luo, W.; Shen, F.; Bommier, C.; Zhu, H.; Ji, X.; Hu, L. Acc. Chem. Res. 2016, 49(2), 231. doi: 10.1021/acs.accounts.5b00482
-
[12]
Lv, Z.-Y.; Feng, R.; Zhao, J.; Fan, H.; Xu, D.; Wu, Q.; Yang, L.-J.; Chen, Q.; Wang, X.-Z.; Hu, Z. Acta Chim. Sinica 2015, 73, 1013(in Chinese). (吕之阳, 冯瑞, 赵进, 范豪, 徐丹, 吴强, 杨立军, 陈强, 王喜章, 胡征, 化学学报, 2015, 73, 1013.)
-
[13]
Ren, T.; Zhuang, Q.-C.; Hao, Y.-W.; Cui, Y.-L. Acta Chim. Sinica 2016, 74, 132(in Chinese). (任彤, 庄全超, 郝玉婉, 崔永丽, 化学学报, 2016, 74, 132.)
-
[14]
Xing, W.; Zhang, Y.; Yan, Z.-F.; Lu, G.-Q. Acta Chim. Sinica 2005, 63, 819(in Chinese). (邢伟, 张颖, 阎子峰, 逯高清, 化学学报, 2005, 63, 819.)
-
[15]
Wen, L.; Liu, C.; Song, R.; Luo, H.-Z; Shi, Y.; Li, F.; Cheng, H. Acta Chim. Sinica 2014, 72, 333(in Chinese). (闻雷, 刘成名, 宋仁升, 罗洪泽, 石颖, 李峰, 成会明, 化学学报, 2014, 72, 333.)
-
[16]
Meng, X.-D.; Zhang, J.-H.; Wang, Y.-Y.; Liu, H. Acta Chim. Sinica 2012, 70, 812(in Chinese). (孟祥德, 张俊红, 王妍妍, 刘海, 化学学报, 2012, 70, 812.)
-
[17]
Yang, S.-B.; Fei, X.-F.; Jiang, N. Acta Chim. Sinica 2009, 67, 1995(in Chinese). (杨绍斌, 费晓飞, 蒋娜, 化学学报, 2009, 67, 1995.)
-
[18]
Dahn, J.-R.; Zheng, T.; Liu, Y.-H.; Xue, J.-S. Science 1995, 270(5236), 590.
-
[19]
Zheng, T.; Liu, Y.; Fuller, E. W.; Tseng, S.; Von Sacken, U.; Dahn, J.-R. J. Electrochem. Soc. 1995, 142(8), 2581.
-
[20]
Liu, Y.-H.; Xue, J.-S.; Zheng, T.; Dahn, J.-R. Carbon 1996, 34(2), 193.
-
[21]
Buiel, E.; Dahn, J.-R. Electrochim. Acta 1999, 45, 121. doi: 10.1016/S0013-4686(99)00198-X
-
[22]
Hashimoto, T.; Yamashita, M.; Kanekiyo, K.; Shiroki, H. Electrochem. Soc. Meet. 1999, 99(2), Abstract no. 157.
-
[23]
Stevens, D.-A.; Dahn, J.-R. J. Electrochem. Soc. 2000, 147(4), 1271.
-
[24]
Stevens, D.-A.; Dahn, J.-R. J. Electrochem. Soc. 2001, 148(8), A803.
-
[25]
Ge, P.; Fouletier, M. Solid State Ionics 1988, 28, 1172.
-
[26]
Cao, Y.; Xiao, L.; Sushko, M.-L.; Wang, W.; Schwenzer, B.; Xiao, J.; Nie, Z.; Saraf, L.-V.; Yang, Z.; Liu, J. Nano Lett. 2012, 12(7), 3783.
-
[27]
David, L.; Singh, G. J. Phys. Chem. C 2014, 118(49), 28401. doi: 10.1021/jp5080847
-
[28]
Wen, Y.; He, K.; Zhu, Y.; Han, F.; Xu, Y.; Matsuda, I.; Ishii, Y.; Cumings, J.; Wang, C. Nat. Commun. 2014, 5, 403.
-
[29]
Wang, Y.; Chou, S.; Liu, H.; Dou, S. Carbon 2013, 57, 202.
-
[30]
Ding, J.; Wang, H.; Li, Z.; Kohandehghan, A.; Cui, K.; Xu, Z.; Zahiri, B.; Tan, X.; Lotfabad, E.-M.; Olsen, B.-C.; Mitlin, D. ACS Nano 2013, 7(12), 11004.
-
[31]
Luo, W.; Jian, Z.; Xing, Z.; Wang, W.; Bommier, C.; Lerner, M.-M.; Ji, X. ACS Central Science 2015, 1(9), 516.
-
[32]
Komaba, S.; Murata, W.; Ishikawa, T.; Yabuuchi, N.; Ozeki, T.; Nakayama, T.; Ogata, A.; Gotoh, K.; Fujiwara, K. Adv. Funct. Mater. 2011, 21(20), 3859. doi: 10.1002/adfm.v21.20
-
[33]
Thomas, P.; Billaud, D. Electrochim. Acta 2002, 47(20), 3303.
-
[34]
Luo, W.; Schardt, J.; Bommier, C.; Wang, B.; Razink, J.; Simonsen, J.; Ji, X. J. Mater. Chem. A 2013, 1(36), 10662. doi: 10.1039/c3ta12389h
-
[35]
Stevens, D.-A.; Dahn, J.-R. J. Electrochem. Soc. 2000, 147(12), 4428.
-
[36]
Fu, L.; Tang, K.; Song, K.; van Aken, P.-A.; Yu, Y.; Maier, J. Nanoscale 2014, 6(3), 1384.
-
[37]
Li, W.; Zeng, L.; Yang, Z.; Gu, L.; Wang, J.; Liu, X.; Cheng, J.; Yu, Y. Nanoscale 2014, 6(6), 693.
-
[38]
Matsuo, Y.; Ueda, K. J. Power Sources 2014, 263, 158. doi: 10.1016/j.jpowsour.2014.04.038
-
[39]
Zhang, G.; Xiong, T.; He, L.; Yan, M.; Zhao, K.; Xu, X.; Mai, L. J. Mater. Sci. 2017, 52(7), 3697. doi: 10.1007/s10853-016-0732-8
-
[40]
Lotfabad, E. M.; Ding, J.; Cui, K.; Kohandehghan, A.; Kalisvaart, W. P.; Hazelton, M.; Mitlin, D. ACS Nano 2014, 8(7), 7115. doi: 10.1021/nn502045y
-
[41]
Ding, J.; Wang, H.; Li, Z.; Cui, K.; Karpuzov, D.; Tan, X.; Kohandehghan, A.; Mitlin, D. Energy Environ. Sci. 2015, 8(3), 941.
-
[42]
Bommier, C.; Surta, T.-W.; Dolgos, M.; Ji, X. Nano Lett. 2015, 15(9), 5888.
-
[43]
Huang, J.; Sumpter, B.-G.; Meunier, V. Chem-Eur J. 2008, 14(22), 6614.
-
[44]
Bommier, C.; Luo, W.; Gao, W.; Greaney, A.; Ma, S.; Ji, X. Carbon 2014, 76, 165.
-
[45]
Zhang, B.; Ghimbeu, C.-M.; Laberty, C.; Vix-Guterl, C.; Tarascon, J. Adv. Energy Mater. 2016, 6(1), 1501588. doi: 10.1002/aenm.201501588
-
[46]
Zhang, S.; Lv, W.; Luo, C.; You, C.; Zhang, J.; Pan, Z.; Kang, F.; Yang, Q. Energy Storage Mater. 2016, 3, 18.
-
[47]
Jache, B.; Adelhelm, P. Angew. Chem. Int. Ed. 2014, 53(38), 10169. doi: 10.1002/anie.201403734
-
[48]
Kim, H.; Hong, J.; Park, Y.; Kim, J.; Hwang, I.; Kang, K. Adv. Funct. Mater. 2015, 25(4), 534. doi: 10.1002/adfm.v25.4
-
[49]
Cohn, A.-P.; Share, K.; Carter, R.; Oakes, L.; Pint, C.-L. Nano Lett. 2016, 16(1), 543.
-
[50]
Kim, H.; Hong, J.; Yoon, G.; Kim, H.; Park, K. Y.; Park, M. S.; Yoon, W.-S.; Kang, K. Energy Environ. Sci. 2015, 8(10), 2963.
-
[51]
Xu, K. Chem. Rev. 2004, 104(10), 4304.
-
[52]
Tobishima, S.; Morimoto, H.; Aoki, M.; Saito, Y.; Inose, T.; Fukumoto, T.; Kuryu, T. Electrochim. Acta 2004, 49(6), 979.
-
[53]
Hasegawa, G.; Kanamori, K.; Kannari, N.; Ozaki, J.; Nakanishi, K.; Abe, T. Chem. Electrochem. 2015, 2(12), 1917.
-
[54]
Wenzel, S.; Hara, T.; Janek, J.; Adelhelm, P. Energy Environ Sci. 2011, 4(9), 3342.
-
[55]
Li, Y.; Hu, Y.; Li, H.; Chen, L.; Huang, X. J. Mater. Chem. A. 2016, 4(1), 96. doi: 10.1039/C5TA08601A
-
[56]
Li, Y.; Hu, Y.; Qi, X.; Rong, X.; Li, H.; Huang, X.; Chen, L. Energy Storage Mater. 2016, 5, 191.
-
[57]
Luo, W.; Bommier, C.; Jian, Z.; Li, X.; Carter, R.; Vail, S.; Lu, Y.; Lee, J.; Ji, X. ACS Appl. Mater. Inter. 2015, 7(4), 2626. doi: 10.1021/am507679x
-
[58]
Shen, F.; Zhu, H.; Luo, W.; Wan, J.; Zhou, L.; Dai, J.; Hu, L. ACS Appl. Mater. Inter. 2015, 7(41), 23291. doi: 10.1021/acsami.5b07583
-
[59]
Li, Y.; Hu, Y.; Titirici, M.; Chen, L.; Huang, X. Adv. Energy Mater. 2016, 6(18), 1600659. doi: 10.1002/aenm.201600659
-
[60]
Thomas, P.; Ghanbaja, J.; Billaud, D. Electrochim Acta 1999, 45(3), 423.
-
[61]
Zhai, Y.; Dou, Y.; Zhao, D.; Fulvio, P.-F.; Mayes, R.-T.; Dai, S. Adv. Mater. 2011, 23(42), 4828.
-
[62]
Stein, A.; Wang, Z.; Fierke, M.-A. Adv. Mater. 2009, 21(3), 265.
-
[63]
Liu, B.; Shioyama, H.; Akita, T.; Xu, Q. J. Am. Chem. Soc. 2008, 130(16), 265.
-
[64]
Rolison, D.-R.; Long, J.-W.; Lytle, J.-C.; Fischer, A.-E.; Rhodes, C.-P.; Mcevoy, T.-M.; Bourga, M.-E.; Lubers, A.-M. Chem. Soc. Rev. 2009, 38(1), 226.
-
[65]
Lee, J.; Kim, J.; Hyeon, T. Adv. Mater. 2006, 18(16), 2073.
-
[66]
Yang, H.-F.; Zhao, D.-Y. J. Mater. Chem. 2005, 15(12), 1217.
-
[67]
Tao, W.; Liu, X.-Y.; Zhao, D.-Y.; Z.; Jiang, Z.-Y. Chem. Phys. Lett. 2004, 389(4-6), 327.
-
[68]
Xin, S.; Guo, Y.; Wan, L. Acc. Chem. Res. 2012, 45(10), 1759. doi: 10.1021/ar300094m
-
[69]
Yao, L. H.; Cao, M. S.; Yang, H. J.; Liu, X. J.; Fang, X. Y.; Yuan, J. Comp. Mater. Sci. 2014, 85, 179.
-
[70]
Zhang, J.; Lv, W.; Tao, Y.; He, Y.; Wang, D.; You, C.; Li, B.; Kang, F.; Yang, Q.-H Energy Storage Mater. 2015, 1, 112.
-
[71]
Li, H.; Shen, F.; Luo, W.; Dai, J.; Han, X.; Chen, Y.; Yao, Y.; Zhu, H.; Fu, K.; Hitz, E.; Hu, L. ACS Appl. Mater. Inter. 2016, 8(3), 2204. doi: 10.1021/acsami.5b10875
-
[72]
Jin, J.; Yu, B.; Shi, Z.; Wang, C.; Chong, C. J. Power Sources 2014, 272, 800. doi: 10.1016/j.jpowsour.2014.08.119
-
[73]
Wu, L.; Buchholz, D.; Vaalma, C.; Giffin, G.-A.; Passerini, S. ChemElectroChem 2016, 3(2), 292.
-
[74]
Shen, F.; Luo, W.; Dai, J.; Yao, Y.; Zhu, M.; Hitz, E.; Tang, Y.; Chen, Y.; Sprenkle, V.-L.; Li, X.; Hu, L. Adv. Energy Mater. 2016, 6(14), 1600377. doi: 10.1002/aenm.201600377
-
[1]
-



图 3 (a~c)钠离子在膨胀石墨中的储存示意图, (d)循环性能和库仑效率, (e)充放电曲线(其中PG指原始石墨, GO指氧化石墨, EG指膨胀石墨)[28]
Figure 3 (a~c) Illustration of Na+ion storage in graphite-based materials; (d) Cycle performance and coulombic efficiency of the sodium cell; (e) Charge/discharge profiles of pristine graphite (PG), graphite oxides (GO), expanded graphite (EG)[28]


图 7 碳分子筛做负极材料的钠离子电池电化学性能: (a)前3圈充放电曲线; (b)前3圈的CV曲线; (c)电流100 mA·g-1时的循环性能; (d)不同电流下的倍率性能[46]
Figure 7 Properties of SIB with the carbon molecule sieve (CMS) anode: (a) Charge/discharge curve of the first three cycles; (b) CV curve of the first three cycles; (c) Cycle performance at the current of 100 mA·g-1; (d) Rate capability at different current densities[46]


图 9 少数片层的石墨烯在醚基电解液中的储钠行为: (a)前5圈充放电曲线; (b)不同倍率充放电曲线对比; (c)倍率性能; (d)循环性能; (e)8000圈循环后的充放电曲线[49]
Figure 9 Sodium storage behavior of few-layer graphene (FLG) electrode with diglyme electrolyte: (a) First five charge/discharge profiles at galvanostatic current density of 0.2 A·g-1. (b) Charge/discharge profiles at galvanostatic current densities ranging from 1 A·g-1to 30 A·g-1 and (c) the corresponding cycling performance, inset shows the linear relation between specific capacity and current density. (d) Extended cycling performance at current density of 12 A·g-1over 8000 cycles with (e) a selected galvanostatic charge/discharge profiles, inset shows that the over potential decreases with cycling[49]

图 10 (a)互相连通的大孔结构的SEM图; (b)样品吸脱附曲线与孔径分布; (c)倍率性能; (d) 0~1.6 V电压区间的比容量[51]
Figure 10 (a) SEM image of the inter connected macroporous structure. (b) Adsorption isotherm and pore size distribution derived from nitrogen adsorption. (c) Rate capability. (d) Contribution of the potentiostatic step at 1.6 V to the total charge capacity upon Na-extraction[51]



图 13 碳分子筛与石墨、活性碳(AC)在钠离子电池中应用的电化学性质对比: (a, d)碳分子筛的CV曲线与前三圈充放电曲线; (b, e)活性碳的CV曲线与前三圈充放电曲线; (c, f)石墨的CV曲线与前三圈充放电曲线[46]
Figure 13 Electrochemical behaviors of CMS and the reference samples. (a, d): CMS; (b, e): AC; (c, f): Graphite. (a~c) CV curves at scan rates of 0.1 mV·s-1 and (d~f) the charge-discharge profiles for the first three cycles at a current density of 100 mA·g-1[46]
-



 下载:
下载:












 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 236
- 文章访问数: 6735
- HTML全文浏览量: 2596
 下载:
下载:
