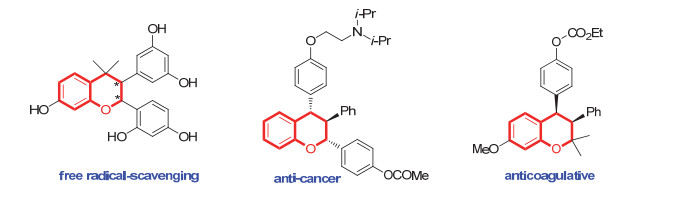
 图 1
含色满骨架的具有代表性的生物活性分子
Figure 1.
Selected bioactive compounds containing chroman core
图 1
含色满骨架的具有代表性的生物活性分子
Figure 1.
Selected bioactive compounds containing chroman core
引用本文:
吴琼, 赵佳佳, 孙斯兵, 屠蔓苏, 石枫. 邻羟基苄醇与邻羟基苯乙烯的催化不对称[4+2]环加成反应——手性色满骨架的立体选择性构建[J]. 化学学报,
2016, 74(7): 576-581.
doi:
10.6023/A16020080

Citation: Wu Qiong, Zhao Jiajia, Sun Sibing, Tu Mansu, Shi Feng. Catalytic Asymmetric[4+2] Cycloaddition of o-Hydroxybenzyl Alcohols with o-Hydroxyl Styrenes: Diastereo-and Enantioselective Construction of Chiral Chroman Scaffold[J]. Acta Chimica Sinica, 2016, 74(7): 576-581. doi: 10.6023/A16020080
Citation: Wu Qiong, Zhao Jiajia, Sun Sibing, Tu Mansu, Shi Feng. Catalytic Asymmetric[4+2] Cycloaddition of o-Hydroxybenzyl Alcohols with o-Hydroxyl Styrenes: Diastereo-and Enantioselective Construction of Chiral Chroman Scaffold[J]. Acta Chimica Sinica, 2016, 74(7): 576-581. doi: 10.6023/A16020080
邻羟基苄醇与邻羟基苯乙烯的催化不对称[4+2]环加成反应——手性色满骨架的立体选择性构建
摘要:
研究了手性磷酸催化下邻羟基苄醇和邻羟基苯乙烯的不对称[4+2]环加成反应,立体选择性地一步构建了手性2,4-二取代四氢色满骨架,该反应具有较高的收率、中等到较高的对映选择性和很好的非对映选择性(最高产率为78%,最高ee值为72%,dr值基本都大于95:5).带有不同取代基的多种邻羟基苄醇和邻羟基苯乙烯均适用于该反应,电子效应对于该反应的对映选择性有一定影响,其中连有供电子基的底物具有更高的反应活性和对映选择性.由邻羟基苄醇原位生成的邻亚甲基苯醌中间体和邻羟基苯乙烯可以同时与催化剂手性磷酸形成双重氢键,对于促进反应的进行和控制该反应的对映选择性起着至关重要的作用.
English
Catalytic Asymmetric[4+2] Cycloaddition of o-Hydroxybenzyl Alcohols with o-Hydroxyl Styrenes: Diastereo-and Enantioselective Construction of Chiral Chroman Scaffold
Abstract:
A chiral phosphoric acid-catalyzed asymmetric[4+2] cycloaddition of o-hydroxyl styrenes with o-quinone methides (o-QMs) generated in situ from o-hydroxybenzyl alcohols has been established. O-Hydroxybenzyl alcohols could transform into o-QM intermediates under the catalysis of chiral phosphoric acid (CPA), which are easily activated by CPA via hydrogen-bonding interaction. On the other hand, o-hydroxyl styrenes could also be activated by CPA via forming a hydrogen bond between the hydroxyl group of styrenes and the phosphoryl oxygen of CPA. So, by selecting o-hydroxybenzyl alcohols as precursors of dienes and o-hydroxyl styrenes as dienophiles under the catalysis of CPA, this catalytic asymmetric[4+2] cycloaddition provided an efficient strategy for constructing enantioenriched chroman framework with two stereogenic centers. A variety of substituted o-hydroxybenzyl alcohols and o-hydroxyl styrenes bearing either electron-donating or electron-withdrawing groups could be applicable to the reaction, delivering chiral chroman derivatives in high yields, considerable enantioselectivities and excellent diastereoselectivities (up to 78% yield, 72% ee, most of examples > 95:5 dr). The electronic nature of the substituents has some effect on the reaction. Namely, the electron-donating groups were beneficial to both the reactivity and the enantioselectivity. Based on the control experiments, it is suggested that the o-hydroxyl styrenes and the o-QM intermediates generated from o-hydroxybenzyl alcohols were simultaneously activated by CPA via forming double hydrogen bonds, thus facilitating the reaction in an enantioselective way. A representative procedure for the enantioselective[4+2] cycloaddition reaction is as following: 1, 2-dichloroethane (1 mL) was added to the mixture of o-hydroxybenzyl alcohols (0.1 mmol), o-hydroxyl styrenes (0.12 mmol), the chiral phosphoric acid (0.005 mmol), and 3 Å molecular sieves (100 mg). After being stirred at 50 ℃ for 12 h, the reaction mixture was filtered to remove the molecular sieves, and the solid powder was washed with ethyl acetate. The resultant solution was concentrated under the reduced pressure to give the residue, which was purified through flash column chromatography on silica gel to afford the pure chiral chroman derivatives.
-
Key words:
- o-hydroxybenzyl alcohol
- / o-hydroxyl styrene
- / [4+2] cycloaddition
- / chroman
- / asymmetric catalysis
-
1 引言
含氧杂环化合物分子骨架广泛存在于许多具有重要生物活性的天然产物或人工合成的药物分子中[1].其中, 色满及其衍生物属于一类重要的含氧杂环化合物, 具有显著的生物活性, 包括治疗乳腺癌、皮肤癌、膀胱癌、前列腺癌, 抗结核分支杆菌[2], 抗幽门螺杆菌以及作为雌激素受体等活性[2](图 1).因此, 色满骨架的构建一直受到有机化学和药物化学工作者的强烈关注.近年来, 大量文献报道了色满类衍生物的合成方法[3].然而, 对映选择性地构建手性色满骨架却报道得比较少.由于手性化合物的两种对映异构体有时会显示出不同甚至迥异的生物活性, 所以对映选择性地合成手性色满衍生物显得尤为重要.
图 1
含色满骨架的具有代表性的生物活性分子
Figure 1.
Selected bioactive compounds containing chroman core
邻羟基苄醇在手性布朗斯特酸(B*-H)催化下可以异构化为邻亚甲基苯醌(o-QM)中间体[4], 它可以与布朗斯特酸之间形成氢键, 通过氢键作用控制反应的对映选择性.近年来, 国内外化学工作者利用邻羟基苄醇的这一反应特点设计了一些新型的催化不对称反应(图 2)[5~7], 其中较为典型的反应有: (1)与吲哚、萘酚等亲核试剂发生亲核取代构建手性三芳基甲烷骨架(图 2, 方程式a)[5]; (2)与1, 3-二羰类化合物或烯胺类化合物发生串联环化反应构建手性色满骨架(图 2, 方程式b)[6]; (3)与富电子烯烃发生氧杂Diels-Alder(D-A)反应构建手性色满骨架(图 2, 方程式c)[7].由此可见, 开发邻羟基苄醇参与的催化不对称[4+2]环加成反应可以合成结构多样的手性色满衍生物, 丰富手性含氧杂环化合物的合成策略.
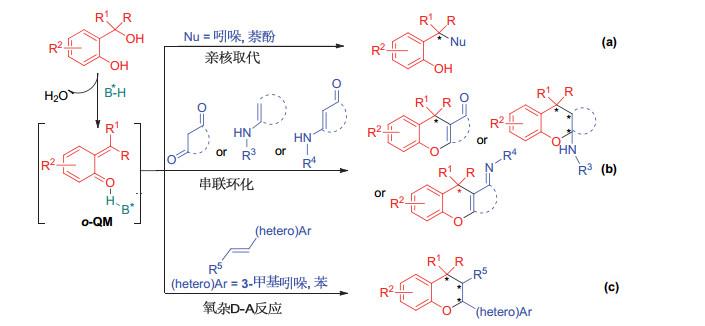 图 2
邻羟基苄醇参与的代表性催化不对称反应
Figure 2.
Representative catalytic asymmetric reactions involving ortho-hydroxybenzyl alcohols
图 2
邻羟基苄醇参与的代表性催化不对称反应
Figure 2.
Representative catalytic asymmetric reactions involving ortho-hydroxybenzyl alcohols
我们课题组利用邻羟基苄醇可以异构化为邻亚甲基苯醌的特性, 使其与3-甲基-2-吲哚烯反应, 在手性磷酸催化下构建了2, 3, 4-三取代的手性四氢色满骨架[7a]; 利用其和达米酮烯胺酮在手性磷酸催化下发生不对称环化反应合成了一系列手性四氢色满衍生物[6e].为了进一步开发邻羟基苄醇参与的催化不对称反应合成具有潜在生物活性的色满衍生物, 我们设计了邻羟基苄醇与邻羟基苯乙烯的催化不对称[4+2]环加成反应, 在手性磷酸(CPA)[8]催化下, 立体选择性地一步构建了2, 4-二取代四氢色满衍生物.值得一提的是, 在我们进行此工作的过程中, Rueping课题组[7b]报道了邻羟基苄醇与苯乙烯在手性膦酰胺催化下的不对称反应, 合成了一系列四氢色满衍生物.由于他们采用的是非活化的苯乙烯, 所以需要采用酸性更强的手性膦酰胺作为催化剂来单侧活化邻亚甲基苯醌中间体.而我们的工作则采用具有活化基团的邻羟基苯乙烯作为富电子烯烃, 通过手性磷酸对两侧底物的双氢键活化作用来控制反应的对映选择性.本文报道我们实验探究所获得的一些结果.
2 结果与讨论
2.1 催化剂及溶剂等因素对反应的影响
首先以氯仿作为溶剂, 在25 ℃下, 以邻羟基苄醇(1a)、邻羟基苯乙烯(2a)为反应底物, 对具有3, 3'-位不同取代基的BINOL骨架衍生的手性磷酸4 (5 mol%)进行筛选(表 1, Entries 1~7).手性磷酸3, 3'-位取代基对反应具有较大的影响, 当3, 3'-位取代基为SiPh3时, 对反应的对映选择性控制最好, 能够以54%的ee获得目标产物3aa(表 1, Entry 1).因此, 我们又将BINOL骨架换成H8-BINOL骨架, 3, 3'-位的取代基依然保持为SiPh3, 尝试该种手性磷酸4h对反应的催化效果.结果发现ee值不但没有提高, 反而稍有下降(表 1, Entry 8).此外, 我们还尝试了手性双磷酸5[9]作为催化剂, ee值也仅为9%, 对该反应的催化活性较差(表 1, Entry 9).所以, 我们仍然采用手性磷酸4a作为最佳的催化剂.
 表 1
反应条件的优化
Table 1.
Optimization of conditions
表 1
反应条件的优化
Table 1.
Optimization of conditions

Entry Cat. Solvent Additives Yieldb/% drc eed/% 1 4a CHCl3 — 36 >95:5 54 2 4b CHCl3 — 70 >95:5 12 3 4c CHCl3 — 64 >95:5 9 4 4d CHCl3 — 74 >95:5 6 5 4e CHCl3 — 15 >95:5 6 6 4f CHCl3 — 24 >95:5 6 7 4g CHCl3 — 58 >95:5 2 8 4h CHCl3 — 37 >95:5 50 9 5 CHCl3 — 29 >95:5 9 10 4a DCE — 58 >95:5 68 11 4a DCE 3 ÅMS 58 >95:5 72 12 e 4a DCE 3 ÅMS 74 >95:5 72 a Unless indicated otherwise, the reaction was carried out in 0.1 mmol scale in a solvent (1.0 mL) at 25 ℃ with additives (100 mg) for 12 h, and the mole ratio of 1a: 2a was 1:1.2. b Isolated yield. c The dr value was determined by 1H NMR. d The enantiomeric excess (ee)value was determined by HPLC. e Performed at 50 ℃. 表 1 反应条件的优化
Table 1. Optimization of conditions确定催化效果最好的手性催化剂以后, 我们又继续对溶剂的种类及用量、分子筛、温度、催化量、当量比进行了细致的筛选(详见支持材料), 发现当溶剂改为1, 2-二氯乙烷(DCE)时可以提高产率和ee值(表 1, Entry 10), 加入3 Å分子筛(100 mg)作为添加剂可以进一步改善ee值(表 1, Entry 11), 在此基础上升温至50 ℃则可以在保持72% ee值的基础上提高产率达74%(表 1, Entry 12).所以, 最终得出最佳的反应条件为(表 1, Entry 12):反应温度为50 ℃, 溶剂为1 mL 1, 2-二氯乙烷, 催化剂为5 mol%手性磷酸4a, 反应物物质的量比为1a:2a=1:1.2, 采用3 Å分子筛(100 mg)作为添加剂.在该最优条件下模板反应的产率为74%, ee值为72%, dr值为>95:5.
2.2 底物的适用范围
2.3 反应机理研究
为了研究该反应的机理, 探明其活化模式, 我们做了相关机理的验证实验(图 4).首先, 我们采用邻甲氧基苄醇1o代替邻羟基苄醇与邻羟基苯乙烯2a在上述最优条件下进行反应, 通过TLC和HPLC监测反应, 发现该反应不能进行; 此外, 我们又采用甲基保护的苯乙烯2f代替邻羟基苯乙烯与邻羟基苄醇1a反应, 发现在相同的条件下也不能发生反应.这两个反应结果表明, 邻羟基苄醇和邻羟基苯乙烯这两类底物中的羟基对于在手性磷酸催化下发生[4+2]环加成反应起着至关重要的作用.这说明邻羟基苄醇原位生成的邻亚甲基苯醌中间体和邻羟基苯乙烯需要和手性磷酸4a形成双重氢键, 通过该双氢键的活化作用, 才能催化反应的进行.
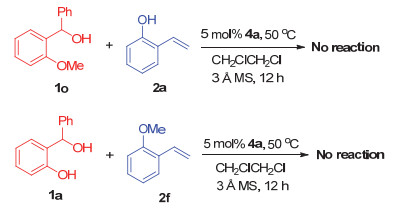 图 4
机理验证实验
Figure 4.
Control experiments
图 4
机理验证实验
Figure 4.
Control experiments
在机理验证实验的基础之上, 我们推测了该反应可能的活化模式(图 5).手性磷酸4a同时与邻羟基苄醇原位生成的邻亚甲基苯醌中间体和邻羟基苯乙烯形成氢键, 促进该[4+2]环加成反应的发生, 从而立体选择性地生成手性色满衍生物3.
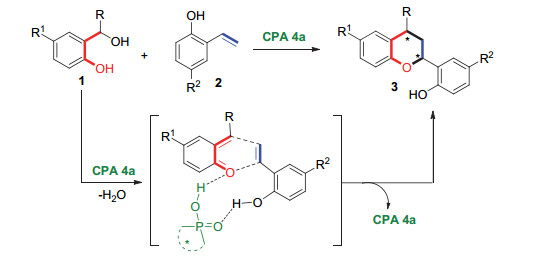 图 5
可能的活化模式
Figure 5.
Suggested activation mode
图 5
可能的活化模式
Figure 5.
Suggested activation mode
产物3的结构通过核磁氢谱、碳谱、红外光谱、高分辨质谱等确定.为了进一步确证产物的结构, 我们还培养出了化合物3ka消旋体的单晶, 并对其进行了单晶X-射线衍射分析(图 6)[10].很遗憾的是, 经过多种尝试, 我们没有培养出手性化合物3的单晶, 所以无法确定产物的绝对构型.但是, 产物3ka的相对构型通过其消旋体的单晶确认为顺式, 即色满环上所连的两个苯基处于该六元含氧杂环的同侧.由于所有产物3的六元含氧杂环中的四个特征氢原子具有相似的峰形和耦合常数, 所以类推它们的相对构型也为顺式.
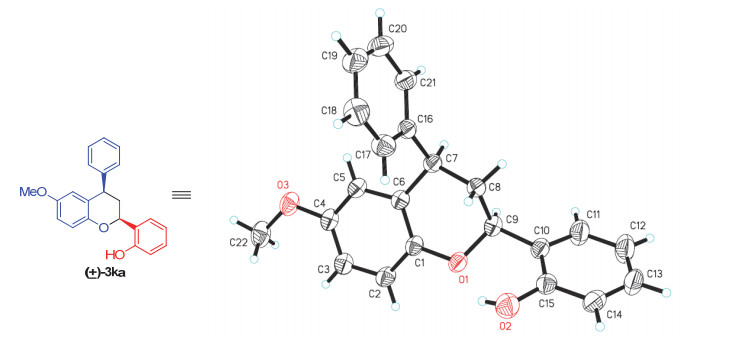 图 6
化合物3ka消旋体的单晶结构
Figure 6.
X-ray structure of racemic compound 3ka
图 6
化合物3ka消旋体的单晶结构
Figure 6.
X-ray structure of racemic compound 3ka
2.2.1 邻羟基苄醇对反应的普适性
通过以上对催化剂的筛选和反应条件的优化, 在最佳反应条件下, 考察具有不同取代基的邻羟基苄醇对反应的普适性, 结果列于表 2中.
表 2
邻羟基苄醇的底物拓展a
Table 2.
Substrate scope of ortho-hydroxybenzyl alcohols

Entry 3 R/R1(1) Yield b/% dr c eed/% 1 3aa Ph/H(1a) 74 >95:5 72 2 3ba p-FC6H4/H(1b) 59 >95:5 68 3 3ca p-ClC6H4/H(1c) 50 >95:5 66 4 3da p-MeC6H4/H(1d) 70 >95:5 70 5 3ea p-MeOC6H4/H(1e) 75 89:11 72 6 3fa m-MeC6H4/H(1f) 69 >95:5 70 7 3ga m-MeOC6H4/H(1g) 59 >95:5 66 8 3ha Ph/4-Me(1h) 78 >95:5 70 9 3ia p-FC6H4/4-MeO(1i) 55 >95:5 66 10 3ja p-ClC6H4/4-MeO(1j) 50 >95:5 66 11 3ka Ph/4-MeO(1k) 70 >95:5 70 12 3la p-MeC6H4/4-MeO(1l) 78 >95:5 68 13 3ma m-MeOC6H4/4-MeO (1m) 61 >95:5 60 14 3na Me/4-MeO (1n) 65 88:12 70 aUnless indicated otherwise, the reaction was carried out in 0.1 mmol scale in CH2ClCH2Cl (1 mL) for 12 h in the presence of 3?MS (100 mg), and the mole ratio of 1: 2a was 1:1.2. bIsolated yield. cThe dr value was determined by 1H NMR. dThe enantiomeric excess (ee) value was determined by HPLC. 表 2 邻羟基苄醇的底物拓展a
Table 2. Substrate scope of ortho-hydroxybenzyl alcohols从表 2的数据可以看出, 该反应对于带有不同取代基R/R1的各种邻羟基苄醇均有较好的适用性.总体而言, 无论是吸电子基还是供电子基, 无论是芳香族取代基还是脂肪族取代基, 均可以取得较高的产率、中等到较高的对映选择性及很好的非对映选择性(最高产率为78%, 最高ee值为72%, dr基本都大于95:5).首先, 我们研究了R基团对该反应的影响, 发现当R基团为芳环时, 芳环上连有供电子基时反应活性较高, 不仅比连有吸电子基时取得的产率更高, 还可以获得更优越的对映选择性(表 2, Entries 4~5 vs. 2~3).此外, 我们还对取代基在芳环上的位置效应也进行了研究, 发现取代基的位置对反应的对映选择性也有一定的影响, 取代基在对位时比取代基在间位时具有更高的对映选择性(表 2, Entries 4~5 vs. 6~7).其次, 我们发现R1基团可以由氢原子更换为甲基或甲氧基, 也能获得中等至较高的对映选择性(表 2, Entries 8~13).值得注意的是, R基团可以由芳基变为甲基(表 2, Entry 14), 产物的产率和对映选择性都较高, 这在邻羟基苄醇参与的催化不对称反应中是比较少见的, 可以大大拓宽邻羟基苄醇的底物适用范围.
2.2.2邻羟基苯乙烯对反应的普适性
为了研究邻羟基苯乙烯对该反应的适用范围, 我们采用了不同的邻羟基苯乙烯作为底物参与反应, 实验所得结果列于表 3.
表 3
邻羟基苯乙烯的底物拓展a
Table 3.
Substrate scope of o-hydroxyl styrenes

Entry 3 R(2) Yield b/% dr c eed/% 1 3aa H(2a) 74 >95:5 72 2 3ab Cl (2b) 60 >95:5 64 3 3ac Br (2c) 62 >95:5 60 4 3ad Me(2d) 74 >95:5 60 5 3ae MeO (2e) 75 >95:5 70 a Unless indicated otherwise, the reaction was carried out in 0.1 mmol scale in CH2ClCH2Cl (1 mL) for 12 h in the presence of 3?MS (100 mg), and the mole ratio of 1a: 2 was 1:1.2. b Isolated yield. c The dr value was determined by 1H NMR. d The enantiomeric excess (ee) value was determined by HPLC. 表 3 邻羟基苯乙烯的底物拓展a
Table 3. Substrate scope of o-hydroxyl styrenes根据表 3的数据可见, 电子效应依然对该反应具有一定的影响, 当邻羟基苯乙烯的R基团为吸电子基时, 反应的活性和对映选择性相对较低.例如, 当R基团为吸电子基Cl和Br时, ee值仅为64%和60%, 产率也仅为60%和62%;而当R基团为供电子基时, 产率和对映选择性均有了一定程度的提高.例如, 当R基团为强供电子基MeO时, ee值可以提高到70%, 产率提高到75% (表 3, Entries 4~5 vs. 2~3).
此外, 我们还初步探索了该反应是否可拓展到其他芳香乙烯体系, 如邻羟基萘乙烯6(图 3).在之前的最佳反应条件下, 邻羟基萘乙烯6也能顺利地与邻羟基苄醇1a发生[4+2]环加成反应, 以较高的产率(70%)和优秀的非对映选择性得到四氢色满化合物7.但是, 产物7的对映选择性却比较低(24% ee), 这说明目前的最佳反应条件对其他芳香乙烯体系并不太适用, 需要通过进一步优化反应条件才有可能获得高的对映选择性.
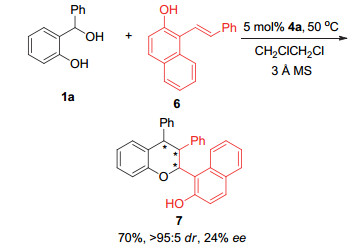 图 3
采用邻羟基萘乙烯6作为底物参与反应
Figure 3.
Reaction using o-hydroxyl naphthalene 6 as a substrate
图 3
采用邻羟基萘乙烯6作为底物参与反应
Figure 3.
Reaction using o-hydroxyl naphthalene 6 as a substrate
3 结论
本文通过手性磷酸催化下邻羟基苄醇和邻羟基苯乙烯的不对称[4+2]环加成反应, 立体选择性地一步构建了2, 4-二取代的手性四氢色满骨架, 反应具有较高的收率、中等到高的对映选择性和很好的非对映选择性(最高产率为78%, 最高ee值为72%, dr值基本都大于95:5).各种取代基的邻羟基苄醇及邻羟基苯乙烯对于该反应均有较好的适用性, 取代基的电子效应对于该反应的对映选择性有一定的影响.通过机理验证实验推测该反应的活化模式为:由邻羟基苄醇原位生成的邻亚甲基苯醌中间体和邻羟基苯乙烯同时和手性磷酸形成双重氢键, 通过该氢键的活化作用来促进反应的进行和控制反应的对映选择性.该反应不仅可以进一步丰富邻羟基苄醇参与的催化不对称反应的研究内容, 还可以为立体选择性地构建手性四氢色满骨架提供一种简洁高效的方法.
-
-
[1]
For some examples, see: (a) Ko, H.-H.; Jin, Y.-J.; Lu, T.-M.; Chen, I.-S. Chem. Biodiversity 2013, 10, 1269; (b) Kumar, S.; Deshpande, S.; Chandra, V.; Kitchlu, S.; Dwivedi, A.; Nayak, V. L.; Konwar, R.; Prabhakar; Yenamandra, S.; Sahu, D. P. Bioorg. Med. Chem. 2009, 17, 6832; (c) Hafez, H. N.; Hegab, M. I.; Ahmed-Farag, I. S.; El-Gazzar, A. B. A. Bioorg. Med. Chem. Lett. 2008, 18, 4538; (d) Poupelin, J. P.; Saint-Ruf, G.; Foussard-Blanpin, O.; Narcisse, G.; Uchida-Ernouf, G.; Lacroix, R. Eur. J. Med. Chem. 1978, 13, 67.
-
[2]
Kumar, S.; Deshpande, S.; Chandra, V.; Kitchlu, S.; Dwivedi, A.; Nayak, V. L.; Konwar, R.; Prabhakar, Y. S.; Sahu, D. P. Bioorg. Med. Chem. 2009, 17, 6832; (b) Sangita; Dwivedi, A.; Prathipati, P.; Ray, S. Med. Chem. Res. 2010, 19, 915.
-
[3]
For some examples, see: (a) Tripathi, S.; Dwivedy, I.; Dhar, J. D.; Dwivedy, A.; Ray, S. Bioorg. Med. Chem. Lett. 1997, 7, 2131; (b) Ferreira, S. B.; da Silva, F. de C.; Pinto, A. C.; Gonzaga, D. T. G.; Ferreira, V. F. J. Heterocycl. Chem. 2009, 46, 1080; (c) Zhang, H.; Zhu, L.; Wang, S.; Yao, Z.-J. J. Org. Chem. 2014, 79, 7063; (d) Yu, S.-Y.; Zhang, H.; Gao, Y.; Mo, L.; Wang, S.; Yao, Z.-J. J. Am. Chem. Soc. 2013, 135, 11402; (e) Enders, D.; Urbanietz, G.; Hahn, R.; Raabe, G. Synthesis 2012, 44, 773.
-
[4]
For some reviews, see: (a) van de Water, R. W.; Pettus, T. R. R. Tetrahedron 2002, 58, 5367; (b) Pathak, T. P.; Sigman, M. S. J. Org. Chem. 2011, 76, 9210; (c) Willis, N. J.; Bray, C. D. Chem.-Eur. J. 2012, 18, 9160; (d) Wang, Z.; Sun, J. Synthesis 2015, 47, 3629; For some enantioselective examples, see: (e) Alden-Danforth, E.; Scerba, M. T.; Lectka, T. Org. Lett. 2008, 10, 4951; (f) Lv, H.; You, L.; Ye, S. Adv. Synth. Catal. 2009, 351, 2822; (g) Pathak, T. P.; Gligorich, K. M.; Welm, B. E.; Sigman, M. S. J. Am. Chem. Soc. 2010, 132, 7870; (h) Luan, Y.; Schaus, S. E. J. Am. Chem. Soc. 2012, 134, 19965; (i) Lv, H.; Jia, W. Q.; Sun, L. H.; Ye, S. Angew. Chem., Int. Ed. 2013, 52, 8607; (j) Izquierdo, J.; Orue, A.; Scheidt, K. A. J. Am. Chem. Soc. 2013, 135, 10634; (k) Wang, Z. B.; Ai, F. J.; Wang, Z.; Zhao, W. X.; Zhu, G. Y.; Lin, Z. Y.; Sun, J. W. J. Am. Chem. Soc. 2015, 137, 383.
-
[5]
Wilcke, D.; Herdtweck, E.; Bach, T. Synlett 2011, 2011, 1235; (b) Zhao, W.; Wang, Z.; Chu, B.; Sun, J. Angew. Chem., Int. Ed. 2015, 54, 1910; (c) Saha, S.; Alamsetti, S. K.; Schneider, C. Chem. Commun. 2015, 51, 1461.
-
[6]
El-Sepelgy, O.; Haseloff, S.; Alamsetti, S. K.; Schneider, C. Angew. Chem., Int. Ed. 2014, 53, 7923; (b) Hsiao, C. C.; Liao, H. H.; Rueping, M. Angew. Chem., Int. Ed. 2014, 53, 13258; (c) Saha, S.; Schneider, C. Chem. Eur. J. 2015, 21, 2348; (d) Saha, S.; Schneider, C. Org. Lett. 2015, 17, 648; (e) Zhao, J.-J.; Zhang, Y.-C.; Xu, M.-M.; Tang, M.; Shi, F. J. Org. Chem. 2015, 80, 10016.
-
[7]
Zhao, J.-J.; Sun, S.-B.; He, S.-H.; Wu, Q.; Shi, F. Angew. Chem., Int. Ed. 2015, 54, 5460; (b) Hsiao, C.-C.; Raja, S.; Liao, H.-H.; Atodiresei, I.; Rueping, M. Angew. Chem., Int. Ed. 2015, 54, 5762. doi: 10.1002/ange.201500215/citedby
-
[8]
For early examples: (a) Akiyama, T.; Itoh, J.; Yokota, K.; Fuchibe, K. Angew. Chem., Int. Ed. 2004, 43, 1566; (b) Uraguchi, D.; Terada, M. J. Am. Chem. Soc. 2004, 126, 5356; For reviews: (c) Akiyama, T. Chem. Rev. 2007, 107, 5744; (d) Terada, M. Chem. Commun. 2008, 35, 4097; (e) Terada, M. Synthesis 2010, 1929; (f) Yu, J.; Shi, F.; Gong, L.-Z. Acc. Chem. Res. 2011, 44, 1156; (g) Parmar, D.; Sugiono, E.; Raja, S.; Rueping, M. Chem. Rev. 2014, 114, 9047; (h) Wu, X.; Li, M.-L.; Gong, L.-Z. Acta Chim. Sinica 2013, 71, 1091. (吴祥, 李明丽, 龚流柱, 化学学报, 2013, 71, 1091.); (i) Wu, H.; He, Y.-P.; Shi, F. Synthesis 2015, 47, 1990; For some selected examples, see: (j) Huang, J.-Z.; Luo, S.-W.; Gong, L.-Z. Acta Chim. Sinica 2013, 71, 879. (黄建洲, 罗时玮, 龚流柱, 化学学报, 2013, 71, 879); (k) Duan, D.; Yin, Q.; Wang, S.; Gu, Q.; You, S. Acta Chim. Sinica 2014, 72, 1001. (段德河, 殷勤, 王守国, 顾庆, 游书力, 化学学报, 2014, 72, 1001.); (l) Shi, L.; Ji, Y.; Huang, W.; Zhou, Y. Acta Chim. Sinica 2014, 72, 820. (时磊, 姬悦, 黄文学, 周永贵, 化学学报, 2014, 72, 820.); (m) Lv, J.; Qin, Y.; Cheng, J.; Luo, S. Acta Chim. Sinica 2014, 72, 809. (吕健, 秦岩, 程津培, 罗三中, 化学学报, 2014, 72, 809.); (n) Wang, S.-G.; You, S.-L. Angew. Chem., Int. Ed. 2014, 53, 2194; (o) Wang, S.-G.; Yin, Q.; Zhuo, C.-X.; You, S.-L. Angew. Chem., Int. Ed. 2015, 54, 647.
-
[9]
Chen, X.-H.; Zhang, W.-Q.; Gong, L.-Z. J. Am. Chem. Soc. 2008, 130, 5652; (b) He, L.; Chen, X.-H.; Wang, D.-N.; Luo, S.-W.; Zhang, W.-Q.; Yu, J.; Ren, L.; Gong, L.-Z. J. Am. Chem. Soc. 2011, 133, 13504.
-
[1]
-

图 2 邻羟基苄醇参与的代表性催化不对称反应
Figure 2 Representative catalytic asymmetric reactions involving ortho-hydroxybenzyl alcohols
表 1 反应条件的优化
Table 1. Optimization of conditions
Entry Cat. Solvent Additives Yieldb/% drc eed/% 1 4a CHCl3 — 36 >95:5 54 2 4b CHCl3 — 70 >95:5 12 3 4c CHCl3 — 64 >95:5 9 4 4d CHCl3 — 74 >95:5 6 5 4e CHCl3 — 15 >95:5 6 6 4f CHCl3 — 24 >95:5 6 7 4g CHCl3 — 58 >95:5 2 8 4h CHCl3 — 37 >95:5 50 9 5 CHCl3 — 29 >95:5 9 10 4a DCE — 58 >95:5 68 11 4a DCE 3 ÅMS 58 >95:5 72 12 e 4a DCE 3 ÅMS 74 >95:5 72 a Unless indicated otherwise, the reaction was carried out in 0.1 mmol scale in a solvent (1.0 mL) at 25 ℃ with additives (100 mg) for 12 h, and the mole ratio of 1a: 2a was 1:1.2. b Isolated yield. c The dr value was determined by 1H NMR. d The enantiomeric excess (ee)value was determined by HPLC. e Performed at 50 ℃.  下载: 导出CSV
下载: 导出CSV
表 2 邻羟基苄醇的底物拓展a
Table 2. Substrate scope of ortho-hydroxybenzyl alcohols
Entry 3 R/R1(1) Yield b/% dr c eed/% 1 3aa Ph/H(1a) 74 >95:5 72 2 3ba p-FC6H4/H(1b) 59 >95:5 68 3 3ca p-ClC6H4/H(1c) 50 >95:5 66 4 3da p-MeC6H4/H(1d) 70 >95:5 70 5 3ea p-MeOC6H4/H(1e) 75 89:11 72 6 3fa m-MeC6H4/H(1f) 69 >95:5 70 7 3ga m-MeOC6H4/H(1g) 59 >95:5 66 8 3ha Ph/4-Me(1h) 78 >95:5 70 9 3ia p-FC6H4/4-MeO(1i) 55 >95:5 66 10 3ja p-ClC6H4/4-MeO(1j) 50 >95:5 66 11 3ka Ph/4-MeO(1k) 70 >95:5 70 12 3la p-MeC6H4/4-MeO(1l) 78 >95:5 68 13 3ma m-MeOC6H4/4-MeO (1m) 61 >95:5 60 14 3na Me/4-MeO (1n) 65 88:12 70 aUnless indicated otherwise, the reaction was carried out in 0.1 mmol scale in CH2ClCH2Cl (1 mL) for 12 h in the presence of 3?MS (100 mg), and the mole ratio of 1: 2a was 1:1.2. bIsolated yield. cThe dr value was determined by 1H NMR. dThe enantiomeric excess (ee) value was determined by HPLC.
下载: 导出CSV
表 3 邻羟基苯乙烯的底物拓展a
Table 3. Substrate scope of o-hydroxyl styrenes
Entry 3 R(2) Yield b/% dr c eed/% 1 3aa H(2a) 74 >95:5 72 2 3ab Cl (2b) 60 >95:5 64 3 3ac Br (2c) 62 >95:5 60 4 3ad Me(2d) 74 >95:5 60 5 3ae MeO (2e) 75 >95:5 70 a Unless indicated otherwise, the reaction was carried out in 0.1 mmol scale in CH2ClCH2Cl (1 mL) for 12 h in the presence of 3?MS (100 mg), and the mole ratio of 1a: 2 was 1:1.2. b Isolated yield. c The dr value was determined by 1H NMR. d The enantiomeric excess (ee) value was determined by HPLC.
下载: 导出CSV
-







 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 0
- 文章访问数: 737
- HTML全文浏览量: 137
 下载:
下载:
