
基于ReaxFF的甲烷无氧转化气相机理研究
English
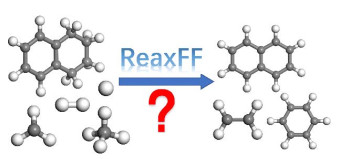
Gas-Phase Mechanism Study of Methane Nonoxidative Conversion by ReaxFF Method
-
-
[1]
Amos, R. D. Mol. Phys. 1979, 38, 33. doi: 10.1080/00268977900101511
-
[2]
Zhan, C. G.; Nichols, J. A.; Dixon, D. A. J. Phys. Chem. A 2003, 107, 4184. doi: 10.1021/jp0225774
-
[3]
Luo, Y. R. Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, 2007; pp. 19–145.
-
[4]
Lunsford, J. H. Catal. Today 2000, 63, 165. doi: 10.1016/S0920-5861(00)00456-9
-
[5]
Schwarz, H. Angew. Chem. Int. Ed. 2011, 50, 10096. doi: 10.1002/anie.201006424
-
[6]
Tang, P.; Zhu, Q. J.; Wu, Z. X.; Ma, D. Energy Environ. Sci. 2014, 7, 2580. doi: 10.1039/c4ee00604f
-
[7]
Weaver, J. F.; Hakanoglu, C.; Antony, A.; Asthagiri, A. Chem. Soc. Rev. 2014, 43, 7536. doi: 10.1039/c3cs60420a
-
[8]
Spivey, J. J.; Hutchings, G. Chem. Soc. Rev. 2014, 43, 792. doi: 10.1039/c3cs60259a
-
[9]
Horn, R.; Schlogl, R. Catal. Lett. 2015, 145, 23. doi: 10.1007/s10562-014-1417-z
-
[10]
Zhao, Z. J.; Chiu, C. C.; Gong, J. L. Chem. Sci. 2015, 6, 4403. doi: 10.1039/c5sc01227a
-
[11]
Olivos-Suarez, A. I.; Szecsenyi, A.; Hensen, E. J. M.; Ruiz-Martinez, J.; Pidko, E. A.; Gascon, J. ACS Catal. 2016, 6, 2965. doi: 10.1021/acscatal.6b00428
-
[12]
Schwach, P.; Pan, X. L.; Bao, X. H. Chem. Rev. 2017, 117, 8497. doi: 10.1021/acs.chemrev.6b00715
-
[13]
Vernon, P. D. F.; Green, M. L. H.; Cheetham, A. K.; Ashcroft, A. T. Catal. Today 1992, 13, 417. doi: 10.1016/0920-5861(92)80167-L
-
[14]
York, A. P. E.; Xiao, T. C.; Green, M. L. H. Top. Catal. 2003, 22, 345. doi: 10.1023/A:1023552709642
-
[15]
Jones, G.; Jakobsen, J. G.; Shim, S. S.; Kleis, J.; Andersson, M. P.; Rossmeisl, J.; Abild-Pedersen, F.; Bligaard, T.; Helveg, S.; Hinnemann, B.; et al. J. Catal. 2008, 259, 147. doi: 10.1016/j.jcat.2008.08.003
-
[16]
Li, D. L.; Nakagawa, Y.; Tomishige, K. Appl. Catal. A 2011, 408, 1. doi: 10.1016/j.apcata.2011.09.018
-
[17]
Pakhare, D.; Spivey, J. Chem. Soc. Rev. 2014, 43, 7813. doi: 10.1039/c3cs60395d
-
[18]
Keller, G. E.; Bhasin, M. M. J. Catal. 1982, 73, 9. doi: 10.1016/0021-9517(82)90075-6
-
[19]
Ito, T.; Wang, J. X.; Lin, C. H.; Lunsford, J. H. J. Am. Chem. Soc. 1985, 107, 5062. doi: 10.1021/ja00304a008
-
[20]
Hutchings, G. J.; Scurrell, M. S.; Woodhouse, J. R. Chem. Soc. Rev. 1989, 18, 251. doi: 10.1039/cs9891800251
-
[21]
Lunsford, J. H. Angew. Chem. Int. Ed. 1995, 34, 970. doi: 10.1002/anie.199509701
-
[22]
Groothaert, M. H.; Smeets, P. J.; Sels, B. F.; Jacobs, P. A.; Schoonheydt, R. A. J. Am. Chem. Soc. 2005, 127, 1394. doi: 10.1021/ja047158u
-
[23]
Palkovits, R.; Antonietti, M.; Kuhn, P.; Thomas, A.; Schuth, F. Angew. Chem. Int. Ed. 2009, 48, 6909. doi: 10.1002/anie.200902009
-
[24]
Kwapien, K.; Paier, J.; Sauer, J.; Geske, M.; Zavyalova, U.; Horn, R.; Schwach, P.; Trunschke, A.; Schlogl, R. Angew. Chem. Int. Ed. 2014, 53, 8774. doi: 10.1002/anie.201310632
-
[25]
Grundner, S.; Markovits, M. A. C.; Li, G.; Tromp, M.; Pidko, E. A.; Hensen, E. J. M.; Jentys, A.; Sanchez-Sanchez, M.; Lercher, J. A. Nat. Commun. 2015, 6, 7546. doi: 10.1038/ncomms8546
-
[26]
Ikuno, T.; Zheng, J.; Vjunov, A.; Sanchez-Sanchez, M.; Ortuno, M. A.; Pahls, D. R.; Fulton, J. L.; Camaioni, D. M.; Li, Z. Y.; Ray, D.; et al. J. Am. Chem. Soc. 2017, 139, 10294. doi: 10.1021/jacs.7b02936
-
[27]
Sushkevich, V. L.; Palagin, D.; Ranocchiari, M.; van Bokhoven, J. A. Science 2017, 356, 523. doi: 10.1126/science.aam9035
-
[28]
Wang, P. W.; Zhao, G. F.; Wang, Y.; Lu, Y. Sci. Adv. 2017, 3, e1603180. doi: 10.1126/sciadv.1603180
-
[29]
Xie, J. J.; Jin, R. X.; Li, A.; Bi, Y. P.; Ruan, Q. S.; Deng, Y. C.; Zhang, Y. J.; Yao, S. Y.; Sankar, G.; Ma, D.; et al. Nat. Catal. 2018, 1, 889. doi: 10.1038/s41929-018-0170-x
-
[30]
Wang, L. S.; Tao, L. X.; Xie, M. S.; Xu, G. F.; Huang, J. S.; Xu, Y. D. Catal. Lett. 1993, 21, 35. doi: 10.1007/BF00767368
-
[31]
Weckhuysen, B. M.; Wang, D. J.; Rosynek, M. P.; Lunsford, J. H. Angew. Chem. Int. Ed. 1997, 36, 2374. doi: 10.1002/anie.199723741
-
[32]
Zhang, C. L.; Li, S. A.; Yuan, Y.; Zhang, W. X.; Wu, T. H.; Lin, L. W. Catal. Lett. 1998, 56, 207. doi: 10.1023/A:1019046104593
-
[33]
Weckhuysen, B. M.; Wang, D. J.; Rosynek, M. P.; Lunsford, J. H. J. Catal. 1998, 175, 338. doi: 10.1006/jcat.1998.2010
-
[34]
Xu, Y. D.; Lin, L. W. Appl. Catal. A 1999, 188, 53. doi: 10.1016/S0926-860x(99)00210-0
-
[35]
Liu, S. T.; Wang, L.; Ohnishi, R.; Ichikawa, M. J. Catal. 1999, 181, 175. doi: 10.1006/jcat.1998.2310
-
[36]
Ma, D.; Shu, Y. Y.; Han, X. W.; Liu, X. M.; Xu, Y. D.; Bao, X. H. J. Phys. Chem. B 2001, 105, 1786. doi: 10.1021/jp002011k
-
[37]
Su, L. L.; Ma, D.; Liu, X. M.; Xu, Y. D.; Bao, X. H. Chin. J. Catal. 2002, 23, 41. doi: 10.3321/j.issn:0253-9837.2002.01.010
-
[38]
Xu, Y. D.; Bao, X. H.; Lin, L. W. J. Catal. 2003, 216, 386. doi: 10.1016/S0021-9517(02)00124-0
-
[39]
Su, L. L.; Liu, L.; Zhuang, J. Q.; Wang, H. X.; Li, Y. G.; Shen, W. J.; Xu, Y. D.; Bao, X. H. Catal. Lett. 2003, 91, 155. doi: 10.1023/B:CATL.0000007149.48132.5a
-
[40]
Ismagilov, Z. R.; Matus, E. V.; Tsikoza, L. T. Energy Environ. Sci. 2008, 1, 526. doi: 10.1039/b810981h
-
[41]
Gao, J.; Zheng, Y. T.; Jehng, J. M.; Tang, Y. D.; Wachs, I. E.; Podkolzin, S. G. Science 2015, 348, 686. doi: 10.1126/science.aaa7048
-
[42]
Sun, C. Y.; Fang, G. Z.; Guo, X. G.; Hu, Y. L.; Ma, S. Q.; Yang, T. H.; Han, J.; Ma, H.; Tan, D. L.; Bao, X. H. J. Energy Chem. 2015, 24, 257. doi: 10.1016/S2095-4956(15)60309-6
-
[43]
Tan, P. L. J. Catal. 2016, 338, 21. doi: 10.1016/j.jcat.2016.01.027
-
[44]
Lai, Y.; Veser, G. Catal. Sci. Technol. 2016, 6, 5440. doi: 10.1039/c5cy02258d
-
[45]
Sun, K. D.; Ginosar, D. M.; He, T.; Zhang, Y. L.; Fan, M. H.; Chen, R. P. Ind. Eng. Chem. Res. 2018, 57, 1768. doi: 10.1021/acs.iecr.7b04707
-
[46]
陈强, 姜利学, 李海方, 陈娇娇, 赵艳霞, 何圣贵.物理化学学报, 2019, 35, 1014. doi: 10.3866/PKU.WHXB201811039Chen, Q.; Jiang, L. X.; Li, H. F.; Chen, J. J.; Zhao, Y. X.; He, S. G. Acta Phys. -Chim. Sin. 2019, 35, 1014. doi: 10.3866/PKU.WHXB201811039
-
[47]
王丹, 丁迅雷, 廖珩璐, 戴佳钰.物理化学学报, 2019, 35, 1005. doi: 10.3866/PKU.WHXB201809006Wang, D.; Ding, X. L.; Liao, H. L.; Dai, J. Y. Acta Phys. -Chim. Sin. 2019, 35, 1005. doi: 10.3866/PKU.WHXB201809006
-
[48]
Guo, X. G.; Fang, G. Z.; Li, G.; Ma, H.; Fan, H. J.; Yu, L.; Ma, C.; Wu, X.; Deng, D. H.; Wei, M. M.; et al. Science 2014, 344, 616. doi: 10.1126/science.1253150
-
[49]
Hao, J. Q.; Schwach, P.; Fang, G. Z.; Guo, X. G.; Zhang, H. L.; Shen, H.; Huang, X.; Eggart, D.; Pan, X. L.; Bao, X. H. ACS Catal. 2019, 9, 9045. doi: 10.1021/acscatal.9b01771
-
[50]
Kim, S. K.; Kim, H. W.; Han, S. J.; Lee, S. W.; Shin, J.; Kim, Y. T. Commun. Chem. 2020, 3, 58. doi: 10.1038/s42004-020-0306-1
-
[51]
Liu, Y.; Liu, J. C.; Li, T. H.; Duan, Z. H.; Zhang, T. Y.; Yan, M.; Li, W. L.; Xiao, H.; Wang, Y. G.; Chang, C. R.; et al. Angew. Chem. Int. Ed. 2020, 59, 18586. doi: 10.1002/anie.202003908
-
[52]
van Duin, A. C. T.; Goddard, W. A.; Islam, M. M.; van Schoot, H.; Trnka, T.; Yakovlev, A. L. ReaxFF, 2017, SCM, Theoretical Chemistry, Vrije Universiteit, Amsterdam, The Netherlands, http://www.scm.com
-
[53]
Martyna, G. J.; Klein, M. L.; Tuckerman, M. J. Chem. Phys. 1992, 97, 2635. doi: 10.1063/1.463940
-
[54]
Chenoweth, K.; van Duin, A. C. T.; Goddard, W. A. J. Phys. Chem. A 2008, 112, 1040. doi: 10.1021/jp709896w
-
[55]
Dontgen, M.; Przybylski-Freund, M. D.; Kroger, L. C.; Kopp, W. A.; Ismail, A. E.; Leonhard, K. J. Chem. Theory Comput. 2015, 11, 2517. doi: 10.1021/acs.jctc.5b00201
-
[56]
Zhao, Y.; Truhlar, D. G. Theor. Chem. Acc. 2008, 120, 215. doi: 10.1007/s00214-007-0401-8
-
[57]
Dunning, T. H. J. Chem. Phys. 1989, 90, 1007. doi: 10.1063/1.456153
-
[58]
Purvis, G. D.; Bartlett, R. J. J. Chem. Phys. 1982, 76, 1910. doi: 10.1063/1.443164
-
[59]
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Petersson, G. A.; Nakatsuji, H.; et al. Gaussian 09, Revision D.01; Gaussian Inc.: Wallingford, CT, 2013.
-
[1]
-

 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 51
- 文章访问数: 2437
- HTML全文浏览量: 573
 下载:
下载:
