
图 1.
p-Co-R催化剂的CO/(H2+CO)-TPD谱图
Figure 1.
CO/(H2+CO)-TPD profiles of p-Co-R catalyst

费托(F-T)合成[1]是合成气(CO+H2)在催化剂的作用下合成液体燃料或化学产品的工艺方法,是实现化石燃料清洁利用和开发的有效途径。费托合成催化剂体系主要包括铁(Fe)基、钌(Ru)基、钴(Co)基和镍(Ni)基等。由于Co基催化剂在费托合成中有反应活性高、水煤气变换反应活性低、高长链烃选择性、低CO2选择性、催化剂寿命长等优点,一直以来是费托合成反应催化剂体系的研究重点[2-5]。CO的活化是费托反应的起始步骤,是生成CHx物种的重要过程,同时也是碳链增长的前提,被认为是F-T合成反应的速率控制步骤[6]。
目前,对于CO解离方式的研究报道主要集中在理论计算方面。Liu等[7]基于第一性原理的动力学理论计算,认为在HCP Co上倾向于直接解离C≡O键,在FCC Co上倾向于以氢助解离的方式进行。Shetty等[8]发现在Co(10-10)晶面上,CO主要是通过直接解离的方式进行活化。Cheng等[9]研究了CO在平坦的Co(0001)表面和台阶状的Co(0001)表面的氢助解离过程。CO在台阶Co(0001)表面进行氢助解离生成HCO需要克服的能垒较低,表明台阶状的Co(0001)表面更有利于CO氢助解离。而Inderwildi等[10]表明在Co(0001)表面,CHx通过HCO和H2CO中间体形成,并不遵循碳化物机理。为了揭示CO加氢的相关机理,实验上采用化学瞬态技术跟踪反应器出口气氛的研究方法也有一定进展。化学瞬态动力学(Chemical Transient Kinetics)是研究大气压下催化反应动力学和相关反应机理的有力工具。1939年Wagner等[11]利用化学瞬态动力学来研究非均相催化反应,研究了催化剂在电化学固态系统中电导率的变化,并被Tamaru[12]用来表征多相催化中催化剂的吸附状态。Athariboroujeny等[13]利用化学瞬态动力学研究了Co-MnOx模型催化剂上CO在常压下的加氢反应,认为在单层极限的覆盖值下,CO的快速解离可能是由于氢的辅助作用和Mn2+的促进作用。Chen等[14]采用稳态同位素瞬态动力学分析(SSITKA)和化学瞬态动力学分析(CTKA)的方法研究了负载型Co基催化剂上CO加氢的反应机理,认为CO直接解离比CO氢助解离更为准确。显然,从以上文献来看,不同的研究团队,从理论计算和实验上对CO在Co基催化剂表面的解离方式给出了不同的研究结果。
众所周知,CO解离是费托合成过程的关键反应步骤[15],其解离路径取决于活性金属的表面结构以及活性位点的类型。目前,尚未从实验角度对特定晶面Co基催化剂的CO吸附能力和解离方式有直观的研究。本研究利用溶剂热法合成了三种暴露不同晶面的Co基催化剂,分别是台阶状(10-11)晶面的p-Co-R催化剂、平面状(0001)晶面的s-Co-R催化剂、锯齿状(11-20)晶面的c-Co-R催化剂。在排除载体、助剂以及颗粒尺寸对F-T合成反应性能影响的前提下,利用化学瞬变反应技术(CTK)研究了常压条件下CO在三种暴露不同晶面催化剂上的加氢反应,对费托合成的初始态动力学行为进行跟踪,并结合其他表征手段来研究CO的解离活化方式。
三种暴露不同晶面的Co基催化剂的制备过程采用Qin等[16]文献中报道的方法。
纳米片状钴催化剂的制备步骤如下:无水乙醇和去离子水各20 mL,4.8 g Co(NO3)2·6H2O和4 g PVP依次加入到上述混合溶液,在室温条件下磁力搅拌30 min后缓慢滴入0.4 mol/L NaOH稀溶液(100 mL)继续搅拌30 min,接着将上述溶液倒入到具有聚四氟乙烯内衬的不锈钢自压反应釜(200 mL)中,密封后在120 ℃条件下反应10 h。自然冷却之后产物经六次离心洗涤后在60 ℃下干燥12 h,最后在马弗炉中以1℃/min的升温速率升到500 ℃,煅烧5 h。最终在5%H2/N2的气氛下进行还原,记为p-Co-R。前期文献[16]表明,该纳米片状的p-Co-R催化剂主要暴露晶面为Co(10-11)。
纳米薄片状钴催化剂的制备步骤如下:在114 mL去离子水中依次加入0.75 g Co(CH3COO)2·4H2O和2.4 g PVP,在室温下搅拌30 min后倒入1 mol/L NaOH溶液(6 mL)继续搅拌20 min,接着将上述溶液倒入到具有聚四氟乙烯内衬的不锈钢自压反应釜(200 mL)中,密封后在180 ℃下反应24 h。自然冷却后,产物经六次离心洗涤后在60 ℃下干燥12 h,最后在马弗炉中以1 ℃/min的升温速率升到500 ℃,煅烧5 h。最终在5%H2/N2的气氛下进行还原,记为s-Co-R。前期文献[16]表明,该纳米薄片状的s-Co-R催化剂主要暴露晶面为Co(0001)。
纳米立方体状钴催化剂的制备步骤如下:向60 mL无水乙醇和40 mL去离子水的混合溶液中加入2.0 g Co(CH3COO)2·4H2O,室温下磁力搅拌20 min后倒入25%氨水(10 mL)继续搅拌20 min,接着将上述溶液倒入到具有聚四氟乙烯内衬的不锈钢自压反应釜(200 mL)中,密封后在170 ℃下反应3 h。室温下自然冷却后,产物经六次离心洗涤后在60 ℃下干燥12 h。最终在5%H2/N2的气氛下进行还原,记为c-Co-R。前期文献[16]表明,该纳米薄片状的c-Co-R催化剂主要暴露晶面为Co(11-20)。
CO/(H2+CO)程序升温脱附是在Micromeritics Atochem 2910吸附仪结合在线质谱测定的,由质谱检测CO信号(m/z=28)。其具体测试步骤分别为:
CO-TPD:首先将样品(100 mg, 60−80目)装入石英管中,通入纯氢以10 ℃/min的速率升温至400 ℃并保持6 h,随后切换成氩气降温至50 ℃,在50 ℃的条件下切换成CO吸附30 min,再切换成氩气吹扫掉样品表面弱吸附和气相的CO,最后在氩气气氛下以10 ℃/min的升温速率升温至800 ℃并记录。
(H2+CO)-TPD:首先将样品(100 mg, 60−80目)装入石英管中,通入纯氢以10 ℃/min的速率升温至400 ℃并保持6 h,随后切换成氩气降温至50 ℃,在50 ℃的条件下切换成H2吸附30 min,接着切换成CO吸附30 min,再切换成氩气吹扫掉样品表面物理吸附的CO,最后在氩气气氛下以10 ℃/min的升温速率升温至800 ℃并记录。
拉曼光谱表征是在上海程方光学有限公司生产的型号为LabRAM HR Evolution拉曼光谱仪上获得的,测试所用的激发光源波长为532 nm。相关步骤如下:通入纯氢以10 ℃/min的速率升温至400 ℃并保持6 h,随后降温至50 ℃切换成合成气升温到250 ℃,分别在0、2、5 h时采集谱图。
样品的原位漫反射红外光谱谱图是通过Thermo Scientific Nicolet iS10光谱仪获得的。相关步骤如下:样品研磨成细粉后装入原位漫反射池内铺平,通入纯氢以10 ℃/min的速率升温至400 ℃并保持6 h,随后降温至30 ℃采集背景,紧接着切换成合成气升温到250 ℃采集谱图。谱图采集过程中扫描64次,分辨率为8 cm−1。
化学瞬态动力学(CTK)的原理是触发反应物进料化学成分的突然变化,并跟踪催化活性相的形成或其清除随时间的变化[13]。从还原气气氛切换到反应气气氛的这一过程称为正向瞬态阶段[17-19],在本实验中从60%H2/40%He切换到60%H2/30%CO/10%Ar,引发费托反应。实验过程中原料气气氛中一直有H2,因此,将该条件下的正向CTK实验也称为富氢条件下的瞬间切气实验,在该阶段能够得到关于催化剂表面物种加氢的相关信息[20]。
化学瞬态反应器内径6 mm,气流通过石英管自上向下流动,石英棉将催化剂固定在恒温区,不锈钢管支撑着催化剂,防止细颗粒催化剂进入管路,本实验中所有的配件都是焊接的且管路设计尽可能短,从而减小因气体在管路滞留可能使得测量过程产生误差。利用在线质谱仪对正向瞬态阶段进行监测以跟踪瞬态实验过程中反应物和产物随着时间的变化。具体实验过程为:称取催化剂200 mg(60−80目)装入反应管,在400 ℃的H2(40 mL/min)气氛下还原6 h,从而保证了Co表面处于准金属状态[13],即保持Co0状态。然后降至反应温度250 ℃,以10 mL/min的稳定流量在60%H2/40%He的气氛下下保持一段时间,通过四通阀切换至60%H2/30%CO/10%Ar,切换时保持常压且流量保持不变。切换后反应物和产物的组成随着时间变化,直至达到稳态,实验过程采用四极质谱跟踪分析。其中,10%Ar作为合成气中的示踪剂,用来表征CO在没有吸附反应情况下的理论响应,记作COtheor。
程序升温脱附是提供CO吸附解离相关信息的重要工具[21],同时也能了解CO与金属催化剂之间的相互作用。图1显示了p-Co-R催化剂的CO/(H2+CO)-TPD中CO的脱附信号。对于p-Co-R催化剂来说,与仅吸附CO的脱附峰相比,当催化剂预吸附H2然后吸附CO时脱附温度降低(77 vs 70 ℃),表明预吸附氢减弱了CO与Co粒子表面的结合能力[22],需要的脱附活化能变低,抑制了p-Co-R催化剂上的CO吸附。CO-金属键的结合能力减弱,吸附的CO中C−O键的强度增强,所以预吸附氢也抑制了p-Co-R催化剂上CO的解离。因此,CO在台阶状(10-11)晶面的p-Co-R催化剂上倾向于直接解离而不是氢助解离,这与文献中理论计算[7,23]结果相一致。
图2显示了s-Co-R催化剂的CO/(H2+CO)-TPD中CO的脱附信号。对于s-Co-R来说,与仅吸附CO相比,预吸附H2然后吸附CO时脱附温度向高温方向偏移(82 vs 100 ℃),且脱附峰面积增加。表明预吸附氢的存在增强了CO与金属活性位点的相互作用,导致CO在s-Co-R催化剂上的低温吸附性能增强。CO−金属键的结合能力增强,C−O键相应地减弱[21],进一步促进了CO的解离和活化。因此,预吸附氢促进了s-Co-R催化剂上CO的解离,表明CO在s-Co-R催化剂上倾向于氢助解离。此外,理论计算表明[24,25],CO在平坦位点容易发生氢助解离,也进一步证明了在平面状(0001)晶面的s-Co-R催化剂上CO倾向于以氢助解离的方式进行活化。
图3显示了c-Co-R催化剂的CO/(H2+CO)-TPD中CO的脱附信号。对于c-Co-R来说,与仅吸附CO相比,预吸附H2然后吸附CO时脱附峰向低温方向偏移(94 vs 86 ℃)。类似于CO在p-Co-R催化剂上的吸附和解离,表明预吸附氢抑制了CO在c-Co-R催化剂上的吸附和解离,CO分子很可能是与H*形成一些中间体HCO*或者COH*。因此,CO在锯齿状的Co(11-20)晶面上倾向于直接解离,与理论计算结果一致[7]。
利用瞬变反应技术考察了所制备的三种不同晶面Co催化剂在250 ℃时CO加氢的初始产物变化。三种催化剂最初在60%H2/40%He的气氛下保持一段时间,使催化剂表面处于H2的动态吸附-脱附平衡,之后经过四通阀瞬间切换到60%H2/30%CO/10%Ar(如图4),反应一段时间后,产物分子开始从催化剂表面脱附。在整个过程中各原料气的流量均保持不变。

图5显示了在250 ℃下p-Co-R催化剂的正向CTKA过程(插图为COtheor与CO归一化响应值的比较),t=0是指Ar在出口流中出现的时刻。从图5插图可以看出,CO在出口出现的时间与COtheor相比延迟大约40 s,在这40 s内碳原子与氧原子在催化剂表面积累,随后在加氢条件下反应生成CH4、CO2和H2O等。Ojeda等[26]通过动力学和理论分析的结合研究,发现CO如果以氢助解离的方式进行活化,那么O将主要以H2O的形式除去;如果CO是直接解离,所形成的化学吸附氧原子将主要以CO2的形式进行脱附。从图5可以看到,p-Co-R催化剂在正向CTKA过程中,只生成了很少的H2O,大量的O以CO2的形式从催化剂表面脱附。因此,在p-Co-R催化剂上CO主要是直接解离。另外,之前报道的文献中表明,p-Co-R催化剂暴露的Co(10-11)晶面具有很多台阶式B5型位点[16],在典型的费托合成反应条件下该表面具有较高的催化活性[7]。p-Co-R催化剂在H2/He气氛时催化剂表面被原子氢所覆盖,由于金属Co表面的H2很容易解离,氢原子的覆盖度较高。当切换到H2/CO/Ar气氛时,CO开始解离,碳原子的覆盖度也增加,CH4作为脱附的第一个产物在反应的初始阶段表现出较高的生成速率。从而进一步证明,在具有B5活性位点的Co(10-11)晶面上CO解离出的碳原子可以快速加氢。因此,在台阶状的Co(10-11)表面上,CO发生了不可逆地化学吸附且以直接解离的方式活化,在该催化剂上CO解离的碳原子有较强的加氢能力。

图6显示了在250 ℃下s-Co-R催化剂的正向CTKA过程(插图为COtheor与CO归一化响应值的比较),t=0是指Ar在出口流中出现的时刻。对于s-Co-R来说,CO在出口出现的时间与COtheor相比延迟大约20 s,表明CO在s-Co-R催化剂表面发生了不可逆吸附。CH4产生了“过冲”现象,这在其他负载型Co基催化剂的CTKA实验当中也观察到了类似的特点[14,27,28]。在250 ℃的实验条件下,CO和CO2的延迟时间相同,CO的急剧增加伴随着产物CO2的同时出现,减弱了表面氧的积累,且两者出现时CH4达到了最大值。这与p-Co-R催化剂上CH4平滑地达到稳定值的现象是截然不同的,这表明吸附氢对CO解离再加氢生成CH4的这一过程在不同结构的催化剂上影响不同的。在s-Co-R催化剂上H2O的响应值高于CO2,因此,CO在s-Co-R催化剂上是以氢助解离的方式进行的。在质谱未检测到CO响应信号的这段时间里,生成的CH4可能是由于吸附的CO发生氢助解离生成HCO或CHO,然后C−O断裂在加氢条件下生成CHx物种,后期CH4的生成主要是大量合成气进入反应器,CO和H2在催化剂表面发生竞争吸附,其表面逐渐被吸附的CO所饱和,该吸附的CO发生加氢反应生成CH4。理论计算表明,吸附的CO或者沉积的碳会诱导催化剂结构重组[29],平面状的Co(0001)面初始活性低,CO氢助解离形成甲酰基,碳原子浓度的增加会使得催化剂表面不稳定,会产生更多的台阶-边缘位点,在这些位点上CO将以较低的活化能进行解离,有利于长链烃的形成,但是在初期阶段主要产生CH4[30],这与在本实验研究条件下s-Co-R催化剂上观察到的现象是一致的。因此,在平面状的Co(0001)表面上,吸附氢的存在促进了该催化剂上CO的吸附,且CO在s-Co-R催化剂上以氢助解离的方式进行活化。

图7显示了在250 ℃下c-Co-R催化剂的正向CTKA过程(插图为COtheor与CO归一化响应值的比较),t=0是指Ar在出口流中出现的时刻。对于c-Co-R来说,CO在出口出现的时间与COtheor相比延迟大约6 s,表明在Co(11-20)晶面上CO的解离能力非常弱,且CO在该催化剂的作用下加氢生成的产物很少,表明在该晶面上CO加氢能力较弱。因此,在富氢气氛下,c-Co-R上CO发生弱解离。

通过反应条件下对三个不同晶面催化剂的CO吸附能力差异的研究,CO在出口出现的时间与COtheor的延迟时间顺序为p-Co-R(40 s)>s-Co-R(20 s)>c-Co-R(6 s),表明在p-Co-R催化剂上CO脱附最慢。
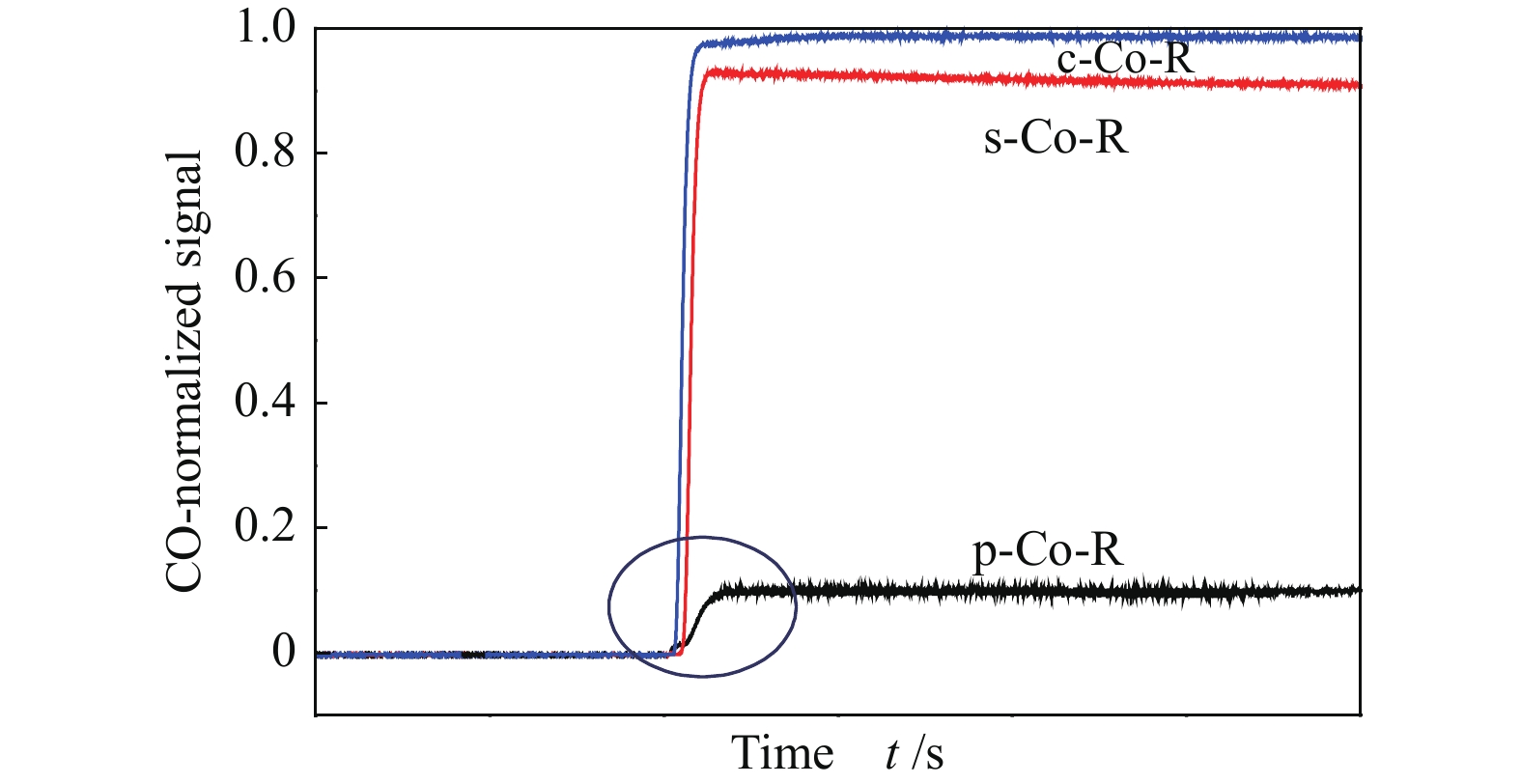
为进一步确认不同晶面Co催化剂的CO吸附行为,将三种催化剂CO的出口流量归一化处理后进行比较,如图8所示。从质谱信号的斜率来看,p-Co-R催化剂的CO出口流量的斜率最小,进一步表明,p-Co-R催化剂上CO脱附速率最慢。就三种催化剂达到稳态的CO消耗值所用时间而言,c-Co-R催化剂达到稳态的CO值所用时间最短,表明暴露Co(11-20)晶面的c-Co-R催化剂上CO达到饱和吸附最快。就CO的响应值来说,c-Co-R催化剂的CO稳态高度最高,意味在该催化剂上反应消耗的CO量最少,CO在该晶面上较难解离和活化,这可能是由于c-Co-R催化剂能够吸附CO的活性位点数量较少造成的。因此,在p-Co-R(10-11)晶面上CO是强吸附的过程,在s-Co-R(0001)和c-Co-R(11-20)晶面上CO是弱吸附的过程,进一步证明了三种催化剂上CO吸附能力强弱顺序为p-Co-R>s-Co-R>c-Co-R。Qin 等[16]研究认为,三种暴露不同晶面的催化剂的CH4选择性顺序为p-Co-R(10-11) < s-Co-R(0001) < c-Co-R(11-20),表明Co(10-11)晶面上吸附的CO最容易解离活化,具有最高的CO加氢能力,与本研究条件得到的结论是一致的。
为了进一步研究CO解离之后的反应路径,对上述三种暴露不同晶面的Co基催化剂进行原位漫反射红外和原位拉曼实验。图9是催化剂p-Co-R、s-Co-R、c-Co-R在250 ℃合成气气氛下各自0、2、5 h下获得的原位拉曼光谱谱图。Raman结果表明,三种催化剂反应一段时间后,均在1340(D峰)和1590 cm−1(G峰)处出现了两个明显的炭特征峰[31,32],但其出现积炭峰的时间显著不同。p-Co-R催化剂反应5 h后才开始出现碳的特征峰,而s-Co-R催化剂在通入合成气的瞬间就检测到了积炭,c-Co-R催化剂在反应2 h后观察到了碳的特征峰。以上拉曼光谱结果说明,本研究条件下,三种不同晶面的Co催化剂具有不同的CO解离和积炭行为。

对以上三种不同晶面Co催化剂进行原位红外光谱分析,如图10所示。所有催化剂均在波数2114 cm−1处,出现了一对明显吸收峰,归属为气相CO的吸收峰,值得注意的是吸收峰的强度有所不同。p-Co-R催化剂在2852、2926和2958 cm−1处出现明显CHx吸收峰,其中,2852、2926 cm−1处的峰归属于CH2物种,2958 cm−1处的峰归属于CH3物种。以上原位漫反射红外光谱结果说明,在该催化剂上CO解离生成的碳物种大量加氢生成CHx,进一步证明了在p-Co-R催化剂上CO解离加氢能力较强。文献报道[33,34],CO解离生成的CHx中间体更容易加氢生成CH4,而不是与非解离CO反应,这与p-Co-R正向CTKA过程中CH4表现出很大的生成速率也是一致的。s-Co-R催化剂在2852、2926和2958 cm−1处的吸收峰强度很弱,表明在s-Co-R催化剂上CO解离加氢生成少量CH2、CH3,大部分是CO的气相双峰(2114、2177 cm−1),结合原位拉曼实验可知,s-Co-R催化剂上CO大量解离为积炭,少量碳物种氢化为CHx。c-Co-R催化剂在2852、2926和2958 cm−1处的CHx吸收峰几乎没有,表明在c-Co-R催化剂上CO解离加氢生成微量CHx。
通过以上分析,基本可以得出CO在三种不同晶面催化剂的解离途径。CO在Co(10-11)晶面解离生成的碳物种部分形成积炭,其余碳物种加氢生成CHx;CO在Co(0001)晶面上大量解离为积炭,少量碳物种氢化为CHx;CO在Co(11-20)晶面上弱解离得到微量的积炭,其余碳物种在氢的存在下生成微量的CHx中间体。

基于以上三种不同晶面Co催化剂的TPD、CTKA、原位IR与原位Raman实验结果,图11为p-Co-R、s-Co-R、c-Co-R催化剂上CO解离行为的示意图。CO在Co(10-11)和Co(11-20)晶面上以直接解离的方式进行活化,直接解离生成碳原子和氧原子,碳原子在加氢反应条件下生成CHx物种,氧原子和碳原子结合生成CO2。Co(0001)晶面上CO以氢助解离的方式进行活化,CO先与表面H反应生成HCO*或者CHO*物种,然后再生成CHx物种和H2O。
采用化学瞬态动力学、程序升温脱附、原位漫反射红外光谱和原位拉曼光谱等表征手段相结合揭示了CO在三种暴露不同晶面的Co基催化剂上费托反应过程中CO解离方式以及解离行为。本研究从实验方面证明了不同晶面Co基催化剂上的CO吸附和解离具有结构敏感性。在氢的存在下,暴露(10-11)晶面的台阶状p-Co-R催化剂吸附CO能力弱但具有较强的CO解离能力,且CO是直接解离,在费托反应条件下生成了大量的CHx和少量的积炭。暴露(0001)晶面的平面状s-Co-R催化剂吸附CO能力弱且CO的解离是以氢助解离方式进行,在费托反应条件下生成少量的CHx和大量的积炭。暴露(11-20)晶面的锯齿状c-Co-R催化剂吸附CO能力最强,导致其CO解离能力最弱,且CO也是直接解离,在费托反应条件下生成了微量的CHx中间体和微量的积炭。
JAHANGIRI H, BENNETT J, MAHJOUBI P, WILSON K, GU S. A review of advanced catalyst development for Fischer-Tropsch synthesis of hydrocarbons from biomass derived syngas[J]. Catal Sci Technol,2014,4(8):2210−2229. doi: 10.1039/C4CY00327F
BEZEMER G L, BITTER J H, KUIPERS H, OOSTERBEEK H, HOLEWIJN J E, XU X D, KAPTEIJN F, VAN DILLEN A J, DE JONG K P. Cobalt particle size effects in the Fischer-Tropsch reaction studied with carbon nanofiber supported catalysts[J]. J Am Chem Soc,2006,128(12):3956−3964. doi: 10.1021/ja058282w
DEN BREEJEN J P, RADSTAKE P B, BEZEMER G L, BITTER J H, HOLMEN A, DE JONG K P. On the origin of the cobalt particle size effects in Fischer-Tropsch catalysis[J]. J Am Chem Soc,2009,131(20):7197−7203. doi: 10.1021/ja901006x
YANG J, TVETEN E Z, CHEN D, HOLMEN A. Understanding the effect of cobalt particle size on Fischer-Tropsch synthesis: Surface species and mechanistic studies by SSITKA and Kinetic Isotope Effect[J]. Langmuir,2010,26(21):16558−16567. doi: 10.1021/la101555u
SAVOST’YANOV A P, YAKOVENKO R E, NAROCHNYI G B, BAKUN V G, SULIMA S L, YAKUBA E S, MITCHENKO S A. Industrial catalyst for the selective Fischer-Tropsch synthesis of long-chain hydrocarbons[J]. Kinet Catal,2017,58(1):81−91. doi: 10.1134/S0023158417010062
CIOBICA I M, KRAMER G J, GE Q, NEUROCK M, VAN SANTEN R A. Mechanisms for chain growth in Fischer-Tropsch synthesis over Ru(0001)[J]. J Catal,2002,212(2):136−144. doi: 10.1006/jcat.2002.3742
Liu J X, Su H Y, Sun D P, Zhang B Y, Li W X. Crystallographic dependence of CO activation on cobalt catalysts: HCP versus FCC[J]. J Am Chem Soc,2013,135(44):16284−16287. doi: 10.1021/ja408521w
SHETTY S, VAN SANTEN R A. Hydrogen induced CO activation on open Ru and Co surfaces[J]. Phys Chem Chem Phys,2010,12(24):6330−6332. doi: 10.1039/b926731j
CHENG J, HU P, ELLIS P, FRENCH S, KELLY G, LOK C M. First-principles study of oxygenates on Co surfaces in Fischer-Tropsch synthesis[J]. J Phys Chem C,2008,112(25):9464−9473. doi: 10.1021/jp802242t
INDERWILDI O R, JENKINS S J, KING D A. Fischer-Tropsch mechanism revisited: Alternative pathways for the production of higher hydrocarbons from synthesis gas[J]. J Phys Chem C,2008,112(5):1305−1307. doi: 10.1021/jp710674q
WAGNER C, HAUFFE Z. Untersuchungen ber den stationären zustand von katalysatoren bei heterogenen reaktionen. II.[J]. Zeitchrift fur elektrochemie und angewandte physikalische chemie,1939,45:409−426.
TAMARU K. Adsorption measurements during surface catalysis[J]. Bull Chem Soc Jpn,1958,31(5):666−667. doi: 10.1246/bcsj.31.666
ATHARIBOROUJENY M, RAUB A, IABLOKOV V, CHENAKIN S, KOVARIK L, KRUSE N. Competing mechanisms in CO hydrogenation over Co-MnOx catalysts[J]. ACS Catal,2019,9(6):5603−5612. doi: 10.1021/acscatal.9b00967
CHEN W, PESTMAN R, ZIJLSTRA B, FILOT I A W, HENSEN E J M. Mechanism of cobalt-catalyzed CO hydrogenation: 1. Methanation[J]. ACS Catal,2017,7(12):8050−8060. doi: 10.1021/acscatal.7b02757
SHETTY S, VAN SANTEN R A. CO dissociation on Ru and Co surfaces: The initial step in the Fischer-Tropsch synthesis[J]. Catal Today,2011,171(1):168−173. doi: 10.1016/j.cattod.2011.04.006
QIN C, HOU B, WANG J, WANG Q, WANG G, YU M, CHEN C, JIA L, LI D. Crystal-plane-dependent Fischer-Tropsch performance of cobalt catalysts[J]. ACS Catal,2018,8(10):9447−9455. doi: 10.1021/acscatal.8b01333
SCHWEICHER J, BUNDHOO A, FRENNET A, KRUSE N, DALY H, MEUNIER F C. DRIFTS/MS studies during chemical transients and SSITKA of the CO/H2 reaction over Co-MgO catalysts[J]. J Phys Chem C,2010,114(5):2248−2255. doi: 10.1021/jp909754w
KRUSE N, SCHWEICHER J, BUNDHOO A, FRENNET A, DE BOCARME T V. Catalytic CO hydrogenation: Mechanism and kinetics from chemical transients at low and atmospheric pressures[J]. Top Catal,2008,48(1/4):145−152. doi: 10.1007/s11244-008-9045-8
SCHWEICHER J, BUNDHOO A, KRUSE N. Hydrocarbon chain lengthening in catalytic CO hydrogenation: Evidence for a CO-Insertion Mechanism[J]. ACS Catal,2012,134(39):16135−16138.
RAUB A, KARROUM H, ATHARIBOROUJENY M, KRUSE N. Chemical transient kinetics in studies of the Fischer-Tropsch reaction and beyond[J]. Catal Lett, 2021, 151: 613−626 .
WANG T, DING Y, XIONG J, YAN L, ZHU H, LU Y, LIN L. Effect of vanadium promotion on activated carbon-supported cobalt catalysts in Fischer-Tropsch synthesis[J]. Catal Lett,2006,107(1/2):47−52. doi: 10.1007/s10562-005-9730-1
FLOTO M E, CIUFO R A, HAN S, MULLINS C B. CO dissociation on model Co/SiO2 catalysts-effect of adsorbed hydrogen[J]. Surf Sci,2021,705:121783. doi: 10.1016/j.susc.2020.121783
ZHANG R, LIU F, WNAG Q, WANG B, LI D. Insight into CHx formation in Fischer-Tropsch synthesis on the hexahedron Co catalyst: Effect of surface structure on the preferential mechanism and existence form[J]. Appl Catal A: Gen,2016,525:76−84. doi: 10.1016/j.apcata.2016.07.007
SU H Y, ZHAO Y H, LIU J X, SUN K J, LI W X. First-principles study of structure sensitivity of chain growth and selectivity in Fischer-Tropsch synthesis using HCP cobalt catalysts[J]. Catal Sci Technol,2017,7(14):2967−2977. doi: 10.1039/C7CY00706J
QI Y, YANG J, CHEN D, HOLMEN A. Recent progresses in understanding of Co-based Fischer-Tropsch catalysis by means of transient kinetic studies and theoretical analysis[J] Catal Lett, 2014, 145(1): 145-161.
OJEDA M, NABAR R, NILEKAR A U, ISHIKAWA A, MAVRIKAKIS M, IGLESIA E. CO activation pathways and the mechanism of Fischer-Tropsch synthesis[J]. J Catal,2010,272(2):287−297. doi: 10.1016/j.jcat.2010.04.012
RALSTON W T, MELAET G, SAEPHAN T, SOMORJAI G A. Evidence of structure sensitivity in the Fischer-Tropsch reaction on model cobalt nanoparticles by time-resolved chemical transient kinetics[J]. Angew Chem Int Ed,2017,56(26):7415−7419. doi: 10.1002/anie.201701186
CHEN W, FILOT I A W, PESTMAN R, HENSEN E J M. Mechanism of cobalt-catalyzed CO hydrogenation: 2. Fischer-Tropsch synthesis[J]. ACS Catal,2017,7(12):8061−8071. doi: 10.1021/acscatal.7b02758
ZHANG X, VAN SANTEN R A, HENSEN E J M. Carbon-induced surface transformations of cobalt[J]. ACS Catal,2015,5(2):596−601. doi: 10.1021/cs501484c
VAN SANTEN R A, MARKVOORT A J, FILOT I A W, GHOURI M M, HENSEN E J M. Mechanism and microkinetics of the Fischer-Tropsch reaction[J]. Phys Chem Chem Phys,2013,15(40):17038. doi: 10.1039/c3cp52506f
NI Z, KANG S, BAI J, LI Y, HUANG Y, WANG Z, QIN H, LI X. Uniformity dispersive, anti-coking core@double-shell-structured Co@SiO2@C: Effect of graphitic carbon modified interior pore-walls on C5+ selectivity in Fischer-Tropsch synthesis[J]. J Colloid Interf Sci,2017,505:325−331. doi: 10.1016/j.jcis.2017.05.096
GUAN H, WANG X, CHEN S, BANDO Y, GOLBERG D. Coaxial Cu-Si@C array electrodes for high-performance lithium ion batteries[J]. Chem Commun,2011,47(44):12098−12100. doi: 10.1039/c1cc15595d
PALOMINO R M, MAGEE J W, LLORCA J, SENANAYAKE S D, WHITE M G. The effect of Fe-Rh alloying on CO hydrogenation to C2+ oxygenates[J]. J Catal,2015,329:87−94. doi: 10.1016/j.jcat.2015.04.033
SU J, MAO W, XU X, YANG Z, LI H, XU J, HAN Y. Kinetic study of higher alcohol synthesis directly from syngas over CoCu/SiO2 catalysts[J]. AIChE J,2014,60(5):1797−1809. doi: 10.1002/aic.14354

图 4 正向瞬变实验示意图
Figure 4 Schematic diagram of gas switching behavior that takes place during a build-up CTK experiment

图 5 p-Co-R催化剂上CO加氢过程中的正向CTK响应曲线
Figure 5 CTK build-up for the CO hydrogenation over p-Co-R catalyst at 250 °Cthe inset provides enlargement of the difference between COtheor and CO

图 6 s-Co-R催化剂上CO加氢过程中的正向CTK响应曲线
Figure 6 CTK build-up for the CO hydrogenation over s-Co-R catalyst at 250 °Cthe inset provide enlargement of the difference between COtheor and CO

图 7 c-Co-R催化剂上CO加氢过程中的正向CTK响应曲线
Figure 7 CTK build-up for the CO hydrogenation over c-Co-R catalyst at 250 °Cthe inset provide enlargement of the difference between COtheor and CO

图 8 p-Co-R、s-Co-R和c-Co-R正向CTK过程中CO信号值比较
Figure 8 Comparison of CO signal during forward CTK over p-Co-R, s-Co-R and c-Co-R catalysts

图 9 p-Co-R、s-Co-R和c-Co-R催化剂费托反应条件下的原位拉曼光谱谱图
Figure 9 In-situ Raman spectra of the catalysts during the F-T reaction

图 10 三种催化剂的原位漫反射红外光谱谱图
Figure 10 In-situ DRIFTS for CO hydrogenation over three catalysts

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:




 下载:
下载:




 下载:
下载: