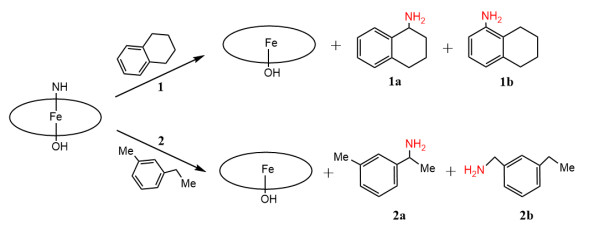
Scheme 1.
Primary amination of C−H bonds catalyzed by cytochrome P411 enzyme

Nitrogen is one of the basic elements present in all living organisms[1]. The primary amine (NH2) is an important, nitrogen-containing functional group that serves as an essential intermediate in the construction of secondary amines, tertiary amines, and heterocycles. Primary amines are widely present in FDA–approved, bestselling drugs such as imatinib, meclizine, clopidogrel, sertraline, rivastigmine, and donepezil among many others[2-5]. The traditional synthesis of primary amines is usually achieved by the reduction of azides or nitriles, the reductive amination of carbonyl compounds[6], or the Buchwald-Hartwig amination of aryl halides[7, 8]. Unfortunately, nitrogen cannot be introduced into natural organic molecules by direct activation of their C–H bonds[9-13]. However, this would be a beneficial protocol as it could convert C−H bonds directly into C−N bonds[14-17]. In recent years, C−H functionalization has emerged as a promising strategy for amine synthesis. For instance, the selective and direct installation of new functional groups into the hydrocarbon framework of organic compounds can greatly simplify amine synthesis, thus reducing waste and promoting sustainable chemical production[18-20]. Some progress has been recently made in protocols providing the primary amination of C(sp2)–H bonds, such as photoredox catalysis[21, 22], electrochemical catalysis[23], and the use of other novel amination reagents and metal catalysts[5, 24-29].
It is well known that enzymes offer numerous advantages to biological catalysis, such as excellent stereoselectivity and high reaction rates. For instance, cytochrome P450 enzymes are hemoproteins that catalyze the hydroxylation of nonactivated C−H bonds with a potentially high degree of stereo- and regioselectivities[30]. However, the primary amination of C(sp3)–H bonds remains a challenge in biology. Notably, Arnold and coworkers successfully designed the cytochrome P411 enzyme through the directed evolution of a cytochrome P450 from Bacillus megaterium, P450BM3. These engineered, iron-heme enzymes can catalyze benzylic and allylic C(sp3)−H aminations with excellent reactivity and regioselectivity[31-33]. Recently, Arnold and coworkers were the first to report the primary amination of C−H bonds catalyzed by NH-bearing iron porphyrin (Fe (Por)(NH)) specie of the P411 enzyme, where tetrahydronaphthalene (1) and 1-(3-methylphenyl)ethane (2)[32] served as the substrates (Scheme 1). In reaction 1, the primary amination of benzylic C−H bond of 1 gives tetrahydronaphthalen-1-amine (1a) as the main product and 1b as a low-yield byproduct. In reaction 2, 1-(3-methylphenyl)ethan-1-amine (2a) is the main product reported, indicating that the primary amination of the secondary C(sp3)–H bond of 2 is more favorable than that of the primary C(sp3)–H bond[32, 34]. However, the active structure of the P411 enzyme and the reaction mechanism of the primary amination of C−H bonds are still unclear, particularly the regioselectivity of the primary amination of different C−H bonds in a substrate.
In this work, we employed the density functional theory (DFT) method to study the active structure of the cytochrome P411 enzyme and the reaction mechanism of the P411-catalyzed primary amination reactions of substrates 1 and 2. Furthermore, we revealed the regioselectivity of the mechanism behind the primary amination of the primary and secondary C(sp3)−H bonds in 2. The electronic characteristics and geometrical structures of the transition states and intermediates of these aminations were also investigated, and reasonable reaction pathways were elucidated.
According to the relevant literature reported by Arnold[32, 33], we used the crystal structure of the variant closely related to P411-B2 (PDB ID: 5UCW) as the initial structure. All the missing hydrogen atoms in the systems were added by the LEaP program of Amber 20 package. The force field for metal and its surrounding amino acids was parameterized by using "MCPB.py" model[35]. The parameters for residue HEM, -NH and substrate were obtained by the parmchk utility from AMBERTools using the general AMBER force field (gaff)[36]. The protonated states of all titratable residues were determined by the PROPKA program at the experimental optimum pH = 7.4[37], and visually inspected using VMD software[38]. To simulate the enzyme-catalyzed environment, the proteins were immersed in A periodic TIP3P tank with a minimum distance of 15 Å from the protein boundary. Then, several Cl-/Na+ ions were added to neutralize the total charges of the systems. The protein in all the simulations were described by the Amber ff14SB force field.
After system setup, first, all the water molecules are minimized while keeping the protein fixed, and then the minimization is performed when the whole system is relaxed. Subsequently, the system was gradually heated from 0 to 300 K under the NVT ensemble for 300 ps with a 1 fs time step[39, 40]. This was followed by equilibrating the density of the systems for 1 ns in NPT ensemble at a target temperature of 300 K and pressure of 1.0 atm. During this procedure, Langevin thermostat with collision frequency of 2 ps-1 and Berendsen barostat with pressure relaxation time of 1 ps were used to maintain the temperature and density of the system. Thereafter, the system was further equilibrated for 4 ns, followed by a productive MD run of 100 ns for the system[41]. The root-mean-squared deviations (RMSD) of the trajectory were calculated for the MD simulation, as shown in Fig. 1. We can see that the whole system remains very stable. The QM region in our QM calculations includes Fe, porphyrin ligand, NH group, and the side chains of serine residues Ser398.
All calculations were performed using the ORCA 3.0.3 software program and UB3LYP functional[42, 43]. We use the ORCA keyword "BrokenSym" to calculate the open-shell state. All geometric optimizations were performed using the Def2-SVP basis set (BS1). Def2-TZVP basis set (BS2) was used for single-point energy calculations. We use SMD model to calculate the solvent effect to correct the single-point energy[44]. Following the conditions set in previous studies exploring enzyme-catalyzed reactions, the SMD solvation model for chlorobenzene was used to simulate a non-polar protein environment, and the dispersion corrections were computed with Grimme's D3BJ method[43]. For all species, the electronic structure was checked by visualizing spin-natural orbitals (SNOs) at the BS1 level and spin-natural orbitals at the BS2 level.
After geometric optimization, the frequencies of all species were calculated to ensure that all the optimized structures had no imaginary frequencies, and that the transition states had only one imaginary frequency with correct vibration direction, from which the zero-point energy was obtained. Intrinsic reaction coordination (IRC) calculations were performed to confirm the relationships among the transition state, the reactant, and the product. In order to facilitate calculation, the porphyrin ligand was modeled as porphine[45], as has been done in similar model system studies. Imitating P450cam, the serine ligand was modeled as −OH[11]. We consider four spin states in total: css (closed-shell singlet), oss (open-shell singlet), triplet and quintet. The optimization of all open-shell singlet structures is carried out based on the initial guess of the triplet state, and then geometric optimization is performed according to the conjecture.
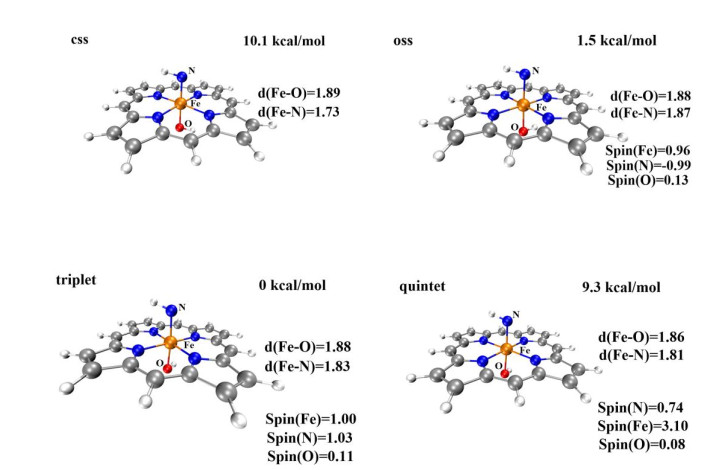
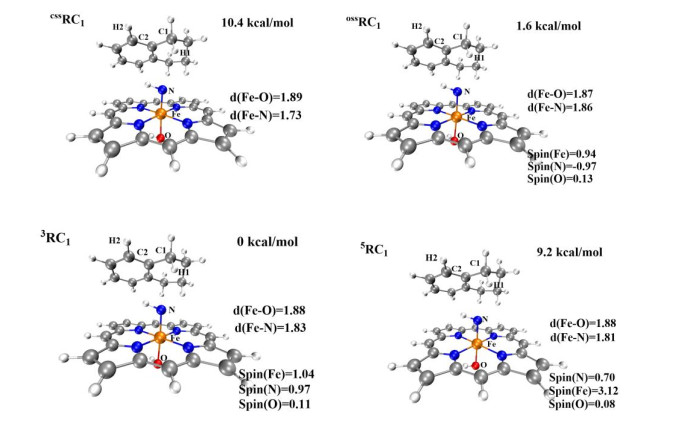
In this section, the geometry and electronic structure of the Fe(Por)(NH) species are investigated. The optimized geometries of Fe(Por)(NH) in four different spin states (css, oss, triplet, and quintet) are presented in Fig. 3. The Fe(Por)(NH) species in the triplet state has the lowest energy among all spin states. The energy level of the open-shell state is only higher than that of the triplet ground state by 1.5 kcal/mol. In contrast, both the quintet state and the closed-shell singlet state lie on energy levels (9.3 and 10.1 kcal/mol, respectively) much higher than that of the triplet ground state. Thus, the primary amination reactions are mainly discussed with respect to the triplet and open-shell singlet state surfaces. The optimized geometries of the Fe(Por)(NH) species in these two states are shown in Fig. 3, where the lengths of Fe–N bonds are 1.83 (triplet state) and 1.87 Å (open-shell singlet state). Based on these values, the Fe–N bonds of Fe(Por)(NH) species in the triplet and open-shell states were determined to be single bonds. The spin densities of the Fe and N atoms in the triplet state are 1.00 and 1.03, respectively. In the open-shell singlet state, the spin density of Fe is 0.96, which is similar to that of its triplet state. However, the spin density of nitrogen in the open-shell singlet state is –0.99 and opposite of its spin density in the triplet state. These calculations indicate that one unit electron might transfer from nitrogen to Fe(Ⅳ), which would result in the formation of a [(por)(–OH)–FeⅢ–N•-–H]- species with an Fe(Ⅲ) metal center and a radical N atom.
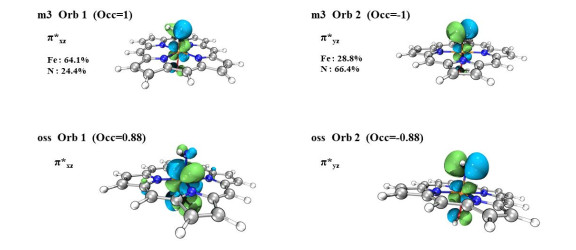
To further understand the electronic configuration of the Fe(Por)(NH) species, we calculated its spin-natural orbitals (SNOs). We found that the triplet state contains two single-occupied orbitals, namely πxz* and πyz* (Fig. 4). The πxz* orbital is composed of the dxz orbitals of Fe atom and the px orbitals of the nitrogen atom and mainly corresponds to the d orbital of Fe atom. The πyz* orbital is composed of the dyz orbitals of the Fe atom and the py orbitals of the nitrogen atom and mainly corresponds to the p orbital of N atom. Furthermore, we used orbital composition analysis with the Mulliken partition to count the contributions of Fe and N atoms in the single-occupied SNOs. The calculations showed that the πyz* orbital (Orb. 2) contributions of the Fe and nitrogen atoms were approximately 28.8% and 66.4%, respectively, which implies that the unpaired electron preferred to occupy the py orbital of the nitrogen atom. Therefore, it would be possible for one unit electron to transfer from the N to the Fe atom, which indicates that a resonance structure of the reactants exists; namely, [(por)(–OH)–FeⅢ–N•-–H]- ↔ [(por)(–OH)–FeⅣ–N2-–H]-. For the open-shell singlet state, the Mulliken partition results show that the contributions of the Fe and N atoms in the πyz* orbital (Orb. 2) were approximately 1.03% and 95.1%, respectively. This result indicates that the unpaired electron is mainly located on the py orbital of the nitrogen atom. However, the biggest difference between the open-shell singlet and triplet states is the single-occupied orbital πyz*, which bears two spin-opposite electrons. The open-shell singlet state exhibits antiferromagnetic coupling between the πyz* orbital which possesses N radical character and the πxz* orbital which owns unpaired electrons on the dxz orbitals of Fe. In contrast, the triplet state exhibited ferromagnetic coupling. Thus, the triplet and open-shell singlet states of Fe(Por)(NH) species are degenerate. Thus, the active structure of the P411 enzyme might exhibit resonance; namely, [(por)(–OH)–FeⅢ–N•-–H]- ↔ [(por)(–OH)–FeⅣ–N2-–H]-.
The amination of the C–H bond of 1 (reaction 1 in Scheme 1) catalyzed by the Fe(Por)(NH) species was then investigated. The optimized geometries of RC1 in four different spin states (css, oss, triplet, and quintet) are shown in Fig. 5. The amination of the C(sp3)−H bond of 1 consists of two main reaction steps: hydrogen-atom abstraction and radical recombination. Hydrogen atom H1 of substrate 1 is the target of the N atom of Fe(Por)(NH) for the abstraction process, and the distance between these two atoms is 3.04 Å. The process of this N atom abstracting H1 was investigated by scanning the energy profile during the acceptance of H1 by the N atom. The calculations indicate that the H1-atom abstraction must tackle a low energy barrier of only 7.1 kcal/mol to reach the transition state of 3TS1, where the N–H1 distance decreases to 1.31 Å. As shown in Fig. 7, the single-occupied πyz* orbital of 3RC1 is mainly distributed on the p orbital of the N atom, which is generally perpendicular to the Fe–N bond and coplanar with the N−H1−C1 plane. As a result, along with the coordination of N−H1 within the H-abstraction process, the radical orbital of the N atom could effectively overlap with the orbital of the C1−H1 bond, which could promote the H-abstraction of 3RC1. Moreover, on the open-shell single state surface, the Fe(Por)(NH) species could undergo a similar H-abstraction process to that of the triplet ground state, except with a slightly higher energy for each structure.
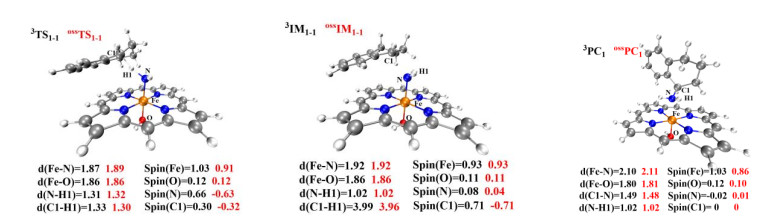
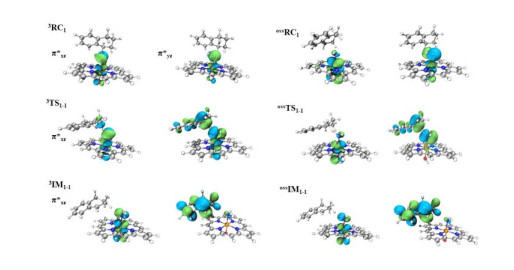
Upon completion of the H-abstraction process, the radical intermediate (3IM1-1) of the [(por)(–OH)–Fe–NH2]- species is generated. The optimized geometry of IM1-1 revealed that the intermediate comprises a coordinated NH2 group and a radial substrate within the cage (Fig. 6). The spin densities of the Fe, N, and C1 atoms changed from 1.04, 0.97, and 0.0 for RC1 to 0.93, 0.08, and 0.71 for IM1-1, respectively. The changes in the spin densities of these atoms from RC1 to IM1-1 indicate that spin-down electron transfers from the C−H1 σ orbital to the N p orbital upon the generation of 3IM1-1 via the H1-atom abstraction process. Subsequently, the radical substrate and Fe(Por)(NH) species in the well-caged should undergo a so-called rebound reaction, which involves the amination of the radical substrate to generate the product. By scanning the energy profile of the process by which the distance between N and C1 along the rebound reaction coordination was shortened, the rebound reaction needed to conquer an energy barrier of only 0.6 kcal/mol to generate the product. Therefore, the H-abstraction is the rate-limiting step of the amination of C(sp3)−H bond of substrate 1. Additionally, this amination process involving open-shell singlet state surface is similar to that involving the triplet ground state, which results in only slightly different energies for each produced structure. The potential energy surface of this reaction is shown in Fig. 8. We also considered another reaction site for the amination of C(sp3)−H bonds, which resulted in a similar reaction process to those presented in the Supporting Information (Fig. S1).
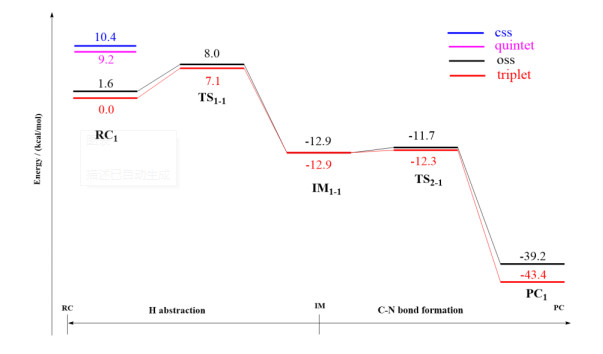
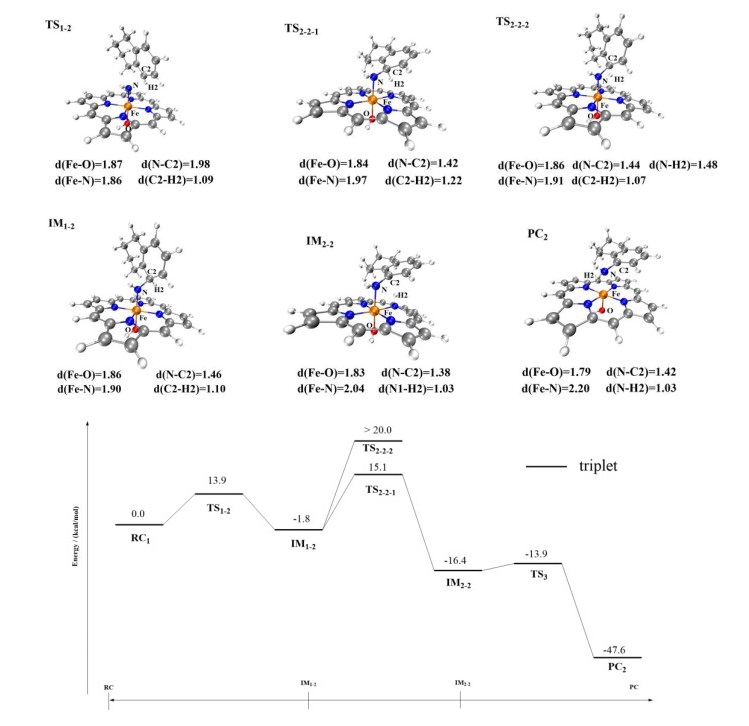
Furthermore, according to the experimental result of Arnold's work[32], the product of C(sp2)−H amination by the P411 enzyme can also be detected in negligible amounts. Thus, we also studied the primary amination of the C(sp2)−H bonds of substrate 1. Since benzene C(sp2)−H bonds are very stable, the direct abstraction of their H atoms is very difficult. We calculated that an energy barrier greater than 30 kcal/mol would need to be overcome to accomplish this (Supporting Information, Fig. S2). Therefore, we investigated another possible reaction pathway for this C(sp2)−H amination reaction involving the assistance of the porphyrin ligand (Fig. 9). Firstly, the N atom directly attacks the C2 atom to form a bridged imine group (energy barrier = 13.9 kcal/mol) to generate IM1-2. Subsequently, the H2 atom of C(sp2)–H bond is abstracted by the coordinated nitrogen. The energy barrier that must be overcome was calculated as 15.1 kcal/mol, which is lower than the energy barrier that must be overcome for the nitrogen to directly abstract the H2 atom (30.6 kcal/mol). Finally, the generated IM2-2 undergoes a rebound reaction with fewer energy barriers and releases the product. Compared with the reaction pathway of C(sp3)−H amination, the reaction of C(sp2)−H amination requires a much higher energy barrier to be conquered than that required by the C(sp3)−H bond amination. These reaction mechanism studies revealed that the H-abstraction is the rate-determining step, and that the P411 enzyme favorably catalyzes the primary amination of the C(sp3)−H bond of tetrahydronaphthalene.
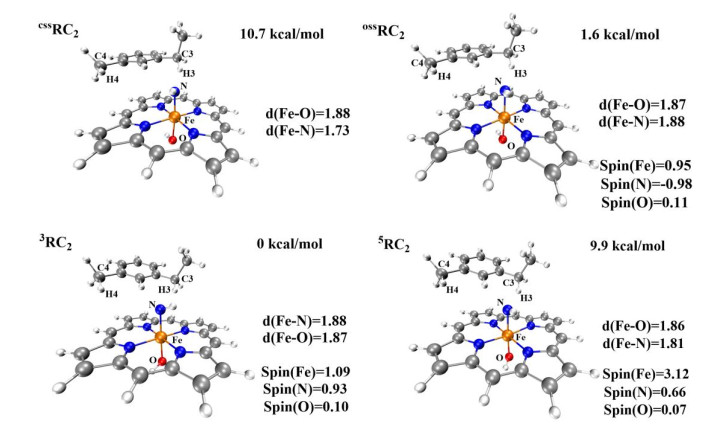
We then investigated the regioselectivity of the P411-enzyme-catalyzed primary amination of different C(sp3)−H bonds (primary and secondary bonds) of 1-(3-methylphenyl) ethane (substrate 2 in Scheme 1). The optimized geometries of RC2 when loading substrate 2 in four different spin states (css, oss, triplet, and quintet) are shown in Fig. 10. As discussed in the previous section, the active site of the triplet ground state and open-shell singlet state of RC2 have the following resonance: [(por)(–OH) FeⅣ–N2-–H]- ↔ [(por)(–OH)–FeⅢ–N•-–H]-. This resonance can activate the C(sp3)−H bond of substrate 2 (Scheme 1). Fig. 11 shows images of the potential energy surfaces of the aminations of primary and secondary C(sp3)−H bonds that take place on the open-shell singlet state and triplet state surfaces, respectively.
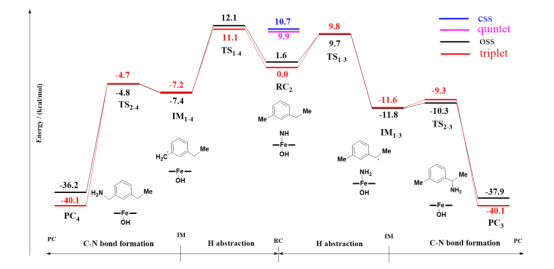
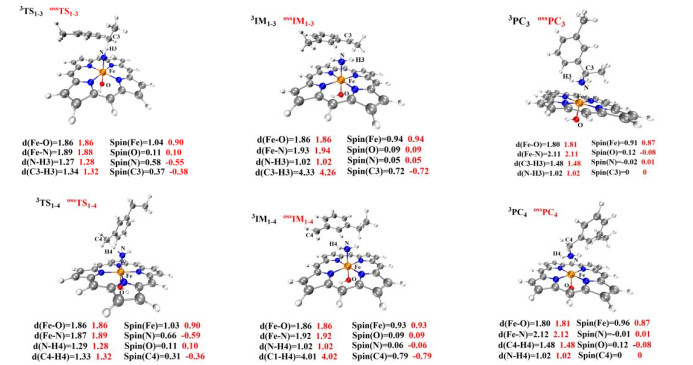
The primary amination of the C(sp3)−H bonds of the Fe(Por)(NH) species should undergo two main reaction steps: H-abstraction and radical recombination. The process by which Fe(Por)(NH) abstracts the H4 atom of 2 was investigated by scanning the energy profile of the N atom as it accepts the H4 atom. The calculations suggested that the abstraction of H4 requires an energy barrier of 11.1 kcal/mol at the triplet state (12.1 kcal/mol at the open-shell single state) to be overcome to generate the radical intermediate of 3IM1-4. Finally, in 3IM1-4, the coordinated NH2 group would recombine with the radical C4 atom with only a slight barrier of 2.5 kcal/mol (2.6 kcal/mol in open-shell singlet) to overcome. In contrast, the H-abstraction step of the primary amination of the secondary C(sp3)−H bonds requires an energy barrier of 9.8 kcal/mol (9.7 kcal/mol in open-shell singlet) to be conquered, which is lower than the barrier that must be overcome for the amination of the primary C(sp3)−H bonds. Moreover, the generated radical intermediate IM1-3 has an energy level of −11.6 kcal/mol (-11.8 kcal/mol in open-shell singlet), which is lower than that of IM1-4 (−7.2 kcal/mol; −7.4 kcal/mol in open-shell singlet). These calculations reveal that the secondary C(sp3)−H bond is more easily activated by the Fe(Por)(NH) species than the primary C(sp3)−H bond. Subsequently, the rebound reaction must overcome only a slight barrier to achieve the rebound of the NH2 group to the radical substrate 2 in IM1-4, resulting in the formation of the product. Additionally, the reaction pathway of Fe(Por)(NH)-catalyzed amination of the secondary C(sp3)−H bonds in the open-shell single state is similar to that in the triplet ground state, with only slightly different energies for each structure. Therefore, the P411 enzyme is energetically favored for aminations of secondary C(sp3)−H bonds.
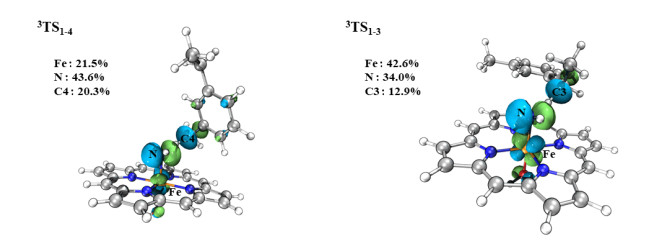
According to our calculations, the H-abstraction step is the rate-determining step of the reaction pathway of the primary amination of C(sp3)−H bonds, which involves an electron transferring from the reactive moiety of the substrate to the Fe(Por)(NH) species. Thus, it can be expected that if the reactive moiety of the substrate is a better electron donor, the barrier of H-abstraction toward that moiety would be decreased. To further understand the reason for the preferred amination of secondary C(sp3)−H bonds, we investigated the SNOs via orbital composition analysis with Mulliken partitions of the primary and secondary C−H moieties of the substrate. As shown in Fig. 13, the single-occupied SNOs of TS1-3 and TS1-4 represent the orbitals interacting when the N atom of Fe(Por)(NH) attacks the primary and secondary C(sp3)−H moieties, respectively of the substrate. For TS1-4, the C4-atom (primary carbon) contribution to the transfer process was 20.3%, while the contributions of N and Fe atoms of the Fe–N center were 43.6% and 21.5%, respectively. For the single-occupied SNOs of TS1-3, the contribution of the C3 atom (secondary carbon) of the substrate to the transfer process was 12.9%, while those of the N and Fe atoms of the Fe–N center were 34.0% and 42.6%, respectively. Upon comparing these results, it is obvious that the contribution of carbon atoms to the single-occupied SNOs of the transition state at the secondary site is lower than that at the primary site. Meanwhile, the higher contribution (42.6~21.5%) of the Fe atom of the Fe–N active center also indicates that the secondary C−H bonds may be more easily reactivated by the Fe(Por)(NH) species than the primary bonds, and the barrier of C−H bond activation should be reduced. This distinguishes the contribution of TS1-3 and TS1-4 to the single-occupied SNOs, indicating that the secondary C(sp3)−H moiety of the substrate exhibits a stronger electron-donor ability than that of the primary C(sp3)−H moiety. As a result, the Fe(Por)(NH) species abstracts the H3 atom from the secondary site of the substrate, which is promoted by the stronger electron-donor ability of the primary site. Therefore, the Fe(Por)(NH) species preferentially catalyzes the primary amination of secondary C(sp3)−H bonds.
In this work, we employed the DFT method to study the electronic structure of the active center of the cytochrome P411 enzyme and the primary amination of C−H bonds catalyzed by its Fe(Por)(NH) species. The calculated spin densities and SNOs indicated the existence of resonance in the reactants; namely, [(por)(–OH) FeⅣ–N2-–H]- ↔ [(por)(–OH)FeⅢ–N•-–H]-. Then, we explored the reactivity of this Fe(Por)(NH) species and revealed the reaction mechanism through which it aminates the C(sp3)−H bonds of substrate tetrahydronaphthalene. The calculated reaction pathway occurring on the triplet ground state surface indicates that the H-abstraction is the rate-determining step of the primary amination. We also found that the energy barrier to be overcome for the subsequent NH2-group rebound reaction to proceed is lower than that of H-abstraction step.
Furthermore, the regioselectivity of Fe(Por)(NH)-catalyzed primary amination of different C(sp3)−H bonds (primary and secondary bonds) was investigated using substrate 1-(3-methylphenyl) ethan. Distinguishing the orbital contribution of single-occupied SNOs in the transition state indicated that the secondary C(sp3)−H moiety of the substrate has a stronger electronic donor ability than that of the primary C(sp3)−H moiety. Therefore, the secondary site of the substrate would be favored for activation by the P411 enzyme. The calculation of the above reactivity and selectivity of the P411 enzyme can provide useful ideas and information for understanding the properties and selectivity of the C−H/C−N bond-activation reactions it catalyzes, as well as those catalyzed by similar enzymes. Our results can also be used for developing and synthesizing new, related catalysts.
Bräse, S. Amino group chemistry: from synthesis to the life sciences. ChemBioChem. 2008, 9, 15093-1509.
Davies, H. M. L.; Morton, D. Collective approach to advancing C-H functionalization. ACS Central Sci. 2017, 3, 936-943. doi: 10.1021/acscentsci.7b00329
Yang, Y.; Cho, I.; Qi, X.; Liu, P.; Arnold, F. H. An enzymatic platform for the asymmetric amination of primary, secondary and tertiary C(sp(3))-H bonds. Nat. Chem. 2019, 11, 987-993. doi: 10.1038/s41557-019-0343-5
Hennessy, E. T.; Betley, T. A. Complex N-heterocycle synthesis via iron-catalyzed, direct C-H bond amination. Science 2013, 340, 591-595. doi: 10.1126/science.1233701
Paudyal, M. P.; Adebesin, A. M.; Burt, S. R.; Ess, D. H.; Ma, Z.; Kurti, L.; Falck, J. R. Dirhodium-catalyzed C-H arene amination using hydroxylamines. Science 2016, 353, 1144-1147. doi: 10.1126/science.aaf8713
Gross, T.; Seayad, A. M.; Ahmad, M.; Beller, M. Synthesis of primary amines: first homogeneously catalyzed reductive amination with ammonia. Org. Lett. 2002, 4, 2055-2058. doi: 10.1021/ol0200605
Hartwig, J. F. Evolution of a fourth generation catalyst for the amination and thioetherification of aryl halides. Accounts Chem. Res. 2008, 41, 1534-1544. doi: 10.1021/ar800098p
Surry, D. S.; Buchwald, S. L. Biaryl phosphane ligands in palladium-catalyzed amination. Angew. Chem., Int. Ed Chem. 2008, 47, 6338-6361. doi: 10.1002/anie.200800497
Dydio, P.; Key, H. M.; Hayashi, H.; Clark, D. S.; Hartwig, J. F. Chemoselective, enzymatic C-H bond amination catalyzed by a cytochrome P450 containing an Ir(Me)-PIX cofactor. J. Am. Chem. Soc. 2017, 139, 1750-1753. doi: 10.1021/jacs.6b11410
Hyster, T. K.; Farwell, C. C.; Buller, A. R.; McIntosh, J. A.; Arnold, F. H. Enzyme-controlled nitrogen-atom transfer enables regiodivergent C-H amination. J. Am. Chem. Soc. 2014, 136, 15505-15508. doi: 10.1021/ja509308v
McIntosh, J. A.; Coelho, P. S.; Farwell, C. C.; Wang, Z. J.; Lewis, J. C.; Brown, T. R.; Arnold, F. H. Enantioselective intramolecular C-H amination catalyzed by engineered cytochrome P450 enzymes in vitro and in vivo. Angew. Chem., Int. Edit. 2013, 52, 9309-9312. doi: 10.1002/anie.201304401
Singh, R.; Bordeaux, M.; Fasan, R. P450-Catalyzed intramolecular sp3 C-H amination with arylsulfonyl azide substrates. ACS Catal. 2014, 4, 546-552. doi: 10.1021/cs400893n
Singh, R.; Kolev, J. N.; Sutera, P. A.; Fasan, R. Enzymatic C(sp3)-H amination: P450-catalyzed conversion of carbonazidates into oxazolidinones. ACS Catal. 2015, 5, 1685-1691. doi: 10.1021/cs5018612
Zalatan, D. N.; Du Bois, J. Metal-catalyzed oxidations of C-H to C-N bonds. C-H activation. Topics in Current Chemistry-Series. 2010, 292, 347-378.
Davies, H. M. L.; Manning, J. R. Catalytic C-H functionalization by metal carbenoid and nitrenoid insertion. Nature 2008, 451, 417-424. doi: 10.1038/nature06485
Collet, F.; Lescot, C.; Dauban, P. Catalytic C-H amination: the stereoselectivity issue. Chem. Soc. Rev. 2011, 40, 1926-1936. doi: 10.1039/c0cs00095g
Jeffrey, J. L.; Sarpong, R. Intramolecular C(sp3)-H amination. Chem. Sci. 2013, 4, 4092-4106. doi: 10.1039/c3sc51420j
Hartwig, J. F. Evolution of C-H bond functionalization from methane to methodology. J. Am. Chem. Soc. 2016, 138, 2-24. doi: 10.1021/jacs.5b08707
Godula, K.; Sames, D. C-H bond functionalization in complex organic synthesis. Science 2006, 312, 67-72. doi: 10.1126/science.1114731
Yamaguchi, J.; Yamaguchi, A. D.; Itami, K. C-H mond functionalization: emerging synthetic tools for natural products and pharmaceuticals. Angew. Chem., Int. Edit. 2012, 51, 8960-9009. doi: 10.1002/anie.201201666
Romero, N. A.; Margrey, K. A.; Tay, N. E. Nicewicz, D. A. Site-selective arene C-H amination via photoredox catalysis. Science 2015, 349, 1326-1330. doi: 10.1126/science.aac9895
Zheng, Y. W.; Chen, B.; Ye, P.; Feng, K.; Wang, W.; Meng, Q. Y.; Wu, L. Z.; Tung, C. H. Photocatalytic hydrogen-evolution cross-couplings: benzene C-H amination and hydroxylation. J. Am. Chem. Soc. 2016, 138, 10080-10083. doi: 10.1021/jacs.6b05498
Morofuji, T.; Shimizu, A.; Yoshida, J. I. Electrochemical C-H amination: synthesis of aromatic primary amines via N-arylpyridinium ions. J. Am. Chem. Soc. 2013, 135, 5000-5003. doi: 10.1021/ja402083e
Peng, J.; Chen, M.; Xie, Z.; Luo, S.; Zhu, Q. Copper-mediated C(sp2)-H amination using TMSN3 as a nitrogen source: redox-neutral access to primary anilines. Org. Chem. Front. 2014, 1, 777-781. doi: 10.1039/C4QO00143E
Tezuka, N.; Shirnojo, K.; Hirano, K.; Komagawa, S.; Yoshida, K.; Wang, C.; Miyamoto, K.; Saito, T.; Takita, R.; Uchiyama, M. Direct hydroxylation and amination of arenes via deprotonative cupration. J. Am. Chem. Soc. 2016, 138, 9166-9171. doi: 10.1021/jacs.6b03855
Legnani, L.; Cerai, G. P.; Morandi, B. Direct and practical synthesis of primary anilines through iron-catalyzed C-H bond amination. ACS Catal. 2016, 6, 8162-8165. doi: 10.1021/acscatal.6b02576
Liu, J.; Wu, K.; Shen, T.; Liang, Y.; Zou, M.; Zhu, Y.; Li, X.; Li, X.; Jiao, N. Fe-catalyzed amination of (hetero)arenes with a redox-active aminating reagent under mild conditions. Chem. Eur. J. 2017, 23, 563-567. doi: 10.1002/chem.201605476
Kim, H.; Heo, J.; Kim, J.; Baik, M. H.; Chang, S. Copper-mediated amination of aryl C-H bonds with the direct use of aqueous ammonia via a disproportionation pathway. J. Am. Chem. Soc. 2018, 140, 14350-14356. doi: 10.1021/jacs.8b08826
Anugu, R. R.; Munnuri, S.; Falck, J. R. Picolinate-directed arene meta-C-H amination via FeCl3 catalysis. J. Am. Chem. Soc. 2020, 142, 5266-5271. doi: 10.1021/jacs.9b13753
Shaik, S.; Cohen, S.; Wang, Y.; Chen, H.; Kumar, D.; Thiel, W. P450 enzymes: their structure, reactivity, and selectivity-modeled by QM/MM calculations. Chem. Rev. 2010, 110, 949-1017. doi: 10.1021/cr900121s
Hyster, T. K.; Arnold, F. H. P450BM3-axial mutations: a gateway to non-natural reactivity. Isr. J. Chem. 2015, 55, 14-20. doi: 10.1002/ijch.201400080
Jia, Z. J.; Gao, S.; Arnold, F. H. Enzymatic primary amination of benzylic and allylic C(sp3)-H bonds. J. Am. Chem. Soc. 2020, 142, 10279-10283. doi: 10.1021/jacs.0c03428
Coelho, P. S.; Wang, Z. J.; Ener, M. E.; Baril, S. A.; Kannan, A.; Arnold, F. H.; Brustad, E. M. A serine-substituted P450 catalyzes highly efficient carbene transfer to olefins in vivo. Nat. Chem. Biol. 2013, 9, 485-U33. doi: 10.1038/nchembio.1278
Wang, J.; Gao, H.; Yang, L.; Gao, Y. Q. Role of engineered iron-haem enzyme in reactivity and stereoselectivity of intermolecular benzylic C-H bond amination. ACS Catal. 2020, 10, 5318-5327. doi: 10.1021/acscatal.0c00248
Li, P.; Merz, K. M. Jr. MCPB. py: a python based metal center parameter builder. J. Chem. Inf. Model. 2016, 56, 599-604. doi: 10.1021/acs.jcim.5b00674
Wang, J. M.; Wolf, R. M.; Caldwell, J. W.; Kollman, P. A.; Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157-1174. doi: 10.1002/jcc.20035
Li, H.; Robertson, A. D.; Jensen, J. H. Very fast empirical prediction and rationalization of protein pKa values. Proteins 2005, 61, 704-721. doi: 10.1002/prot.20660
Humphrey, W.; Dalke, A.; Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 1996, 14, 33-38. doi: 10.1016/0263-7855(96)00018-5
Izaguirre, J. A.; Catarello, D. P.; Wozniak, J. M.; Skeel, R. D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090-2098. doi: 10.1063/1.1332996
Berendsen, H. J. C.; Postma, J. P. M.; Vangunsteren, W. F.; Dinola, A.; Haak, J. R. Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684-3690. doi: 10.1063/1.448118
Shahrokh, K.; Orendt, A.; Yost, G. S.; Cheatham, T. E. Ⅲ. Quantum mechanically derived AMBER-compatible heme parameters for various states of the cytochrome P450 catalytic cycle. J. Comput. Chem. 2012, 33, 119-133. doi: 10.1002/jcc.21922
Neese, F. The ORCA program system. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2012, 2, 73-78. doi: 10.1002/wcms.81
Ahmad, F.; Alam, M. J.; Alam, M.; Azaz, S.; Parveen, M.; Park, S.; Ahmad, S. Synthesis, spectroscopic, computational (DFT/B3LYP), AChE inhibition and antioxidant studies of imidazole derivative. J. Mol. Struct. 2018, 1151, 327-342. doi: 10.1016/j.molstruc.2017.09.056
Marenich, A. V.; Cramer, C. J.; Truhlar, D. G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378-6396. doi: 10.1021/jp810292n
Li, X.; Dong, L.; Liu, Y. Theoretical study of iron porphyrin nitrene: formation mechanism, electronic nature, and intermolecular C-H Amination. Inorg. Chem. 2020, 59, 1622-1632. doi: 10.1021/acs.inorgchem.9b02216

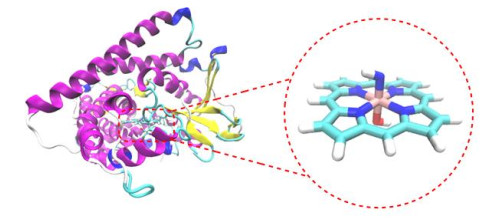
Figure 2 Crystal structure of a variant (PDB ID: 5UCW) closely related to P411-B2 and simplified model used in our calculations

Figure 4 Orbital composition analysis with Mulliken partition of the open-shell singletand triplet states of the Fe(Por)(NH) species

Figure 6 Optimized structures of TS, IM and PC. Some important bond lengths and spin densities f or the open-shell singlet and triplet states are listed. All distances are given in angstroms

Figure 7 Spin-natural orbitals (SNOs) of the open-shell singlet state and triplet state RC1, TS1-1, and IM1-1. All orbitals were computed at the BS2 level of theory and are shown with their occupancies

Figure 11 Potential energy surfaces for the primary aminations of primary (left side of central axis) and secondary (right side of central axis) C(sp3)−H bonds

Figure 12 Geometric optimization of the structures of TS, IM and PC of primary and secondary C(sp3)−H bonds. Some important bond lengths and spin densities for the open-shell singlet and triplet states are provided. All distances are given in angstroms

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载: