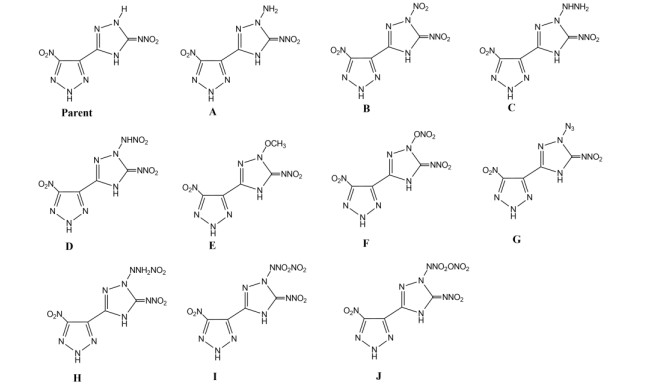
Figure 1.
Derivatives of 4-nitro-5-(5-nitroimino-1, 2, 4-triazol-3-yl)-2H-1, 2, 3-triazolate designed in this paper
Theoretical Study on the Nitrogen-rich Derivatives Based on 1, 2, 4-Triazole and 1, 2, 3-Triazole Rings: an Extended Family of Power Performance Energetic Materials
Jing-Xian JIA , Yu PANG , Jing YANG , Min-Xian LI , Xiang-Jun MENG , Xiao-Zhen GAO , Li-Hua LIU , Meng-Na LIU

High energy density materials (HEDCs) are extensively used in the fields of rocket propulsion systems, gas generators, and explosives[1-10]. It's known that HEDCs should exhibit excellent detonation performance and favorable stability or low sensitivity. It is a pity that the requirements of low sensitivity and high energy are inconsistent with each other, which makes the development of new HEDCs a challenge[11]. Recently, conventional explosives including octahydro-1, 3, 5, 7-tetranitro-1, 3, 5, 7-tetrazocine (HMX) and cyclo-1, 3, 5-trimethylene-2, 4, 6-trinitr-amine (RDX) are widely used as a secondary explosive in the military. However, these typical explosives face similar environmental pollution problems in terms of composition and decomposition products[12]. Therefore, developing green decomposition products as a substitute for classic explosives is an exceedingly good choice. According to most studies, nitrogen-enriched compounds are the choice for synthesizing modern HEDMs as they fulfill most of the requirements, such as high positive heat of formation and high density, while generating environmentally friendly nitrogen gas upon detonation. The molecular framework of nitrogen-enriched compound consists mainly of nitrogen-based heterocyclic rings, such as triazine, tetrazine, triazole, tetrazolium, and other heterocyclic rings[13-15].
Many triazole derivatives have been studied extensively since triazole was first described[16-18]. Meanwhile, extensive efforts have been made to develop new HEDCs based on triazole in heterocyclic energetic materials due to their high and positive heat of formation. Nowadays, Yang research group conduct the study on the synthesis of different neutral compounds consisting of 1, 2, 3-2H-triazole and 1, 2, 4-triazole rings carrying energetic moieties like amino, nitroimino, nitro as well as azo, but unfortunately their detonation performances need to be improved[19]. Thus, we choose the promising 4-nitro-5-(5-nitroimino-1, 2, 4-triazol-3-yl)-2H-1, 2, 3-triazolate in these neutral compounds as the starting material to develop new high energy density materials by the introduction of popular high-energy group, such as -NO2, -NH2, -NHNO2, -NHNH2, -N(NO2NH2) and -N3 groups. Based on density functional theory (DFT), we designed ten molecules (Fig. 1) and evaluated their detonation performance, electronic properties and sensitivity. We sincerely hope that the exploration and calculation of triazole bicyclic derivatives will provide useful information for the development of HEDCs in the future.
The Gaussian 09 program was used to process molecular structure and orbital calculations. The DFT-B3LYP methods with the 6-311G** basis set was adopted for structure optimization[20-22]. The normal mode analysis for each structure gave true local energy minima on the potential energy surfaces with no imaginary frequencies. Heat of formation (HOF) is one significant parameter to evaluate the property of energetic compounds. The isodesmic reaction has been regard as the appropriate approach to study materials fields, especially for the high energy density materials[23-27]. Designed reactions in this work can be written as follows:
|
|
(1) |
where R is NH2, NO2, NHNH2, NHNO2, OCH3, ONO2, N3, NNH2NO2, NNO2NO2 and NNO2ONO2 (Fig. 1). For isodesmic reactions, gas-phase HOF at 298 K can be calculated from the following equation:
|
|
(2) |
where
|
|
(3) |
|
|
(4) |
where (SA) is the molecular surface area for this structure, σtot2 is interpreted as an indicator of the variability of the electrostatic potential on the molecular surface, and ν is described as showing the degree of balance between the positive and negative potentials on the molecular surface.
The significant parameters of an explosive, including the detonation velocity and pressure, were determined by the widely used Empirical Kamlet-Jacob (K-J) equations based on the calculated the enthalpy of formation and density[30]:
|
|
(5) |
|
|
(6) |
where D is the detonation velocity (km/s), P the detonation pressure (GPa), N the moles of gaseous detonation products per gram of explosives, and
|
|
(7) |
 DownLoad:
CSV
DownLoad:
CSV
| Parameters | Explosives components conditions | ||
| c≥2a+b/2 | 2a+b/2 > c≥b/2 | b/2 > c | |
| (b+2c+2d)/4M | (b+2c+2d)/4M | (b+d)/2M | |
| 4M/(b+2c+2d) | (56d+88c-8b)/(b+2c+2d) | (2b+28d+32c)/(b+d) | |
| Q*10-3 | (28.9b+94.05a +0.239 |
[28.9b+94.05(c/2-b/4) +0.239 |
(57.8c+0.239 |
M is the molecular weight of compound. α2, β2 and γ are coefficients[31].
Impact sensitivities (H50) were also estimated as it is an important parameter for predicting the safety of an energetic material during use or storage. For all compounds, impact sensitivity was calculated by a simple method recommended by Pospíšil et al.[32].
|
|
(8) |
Another indicator of explosives, oxygen balance (OB100), is a parameter used to describe the degree of which an explosive can be oxidized. For a compound with molecular formula CaHbOcNd, the oxygen balance can be represented as equation (9)[33].
|
|
(9) |
Here
The heats of formation (HOF), frequently referred to be indicative of the "energy content" of high energetic compounds, is substantial for calculating the detonation performances of energetic compounds. In this work, for convenient discussion, all derivatives are set as derivatives A~L displayed in Fig. 1. Table 2 presents the calculated total energies (E0, a. u), zero-point energies (ZPE, a. u), values of thermal correction (HT, a. u) and HOF (kJ/mol) of the derivatives. All compounds possess positive HOFS in the range of 137.86 to 996.11 kJ/mol. At the same time, the values of all HOFs are significantly higher than that of RDX (95.14 kJ/mol) and 1, 3, 5-triamino-2, 4, 6-trinitrobenzene (TATB = 102.67 kJ/mol)[34]. With the presence of a large number of N–N or C–N bonds in these heterocyclic compounds, especially the two nitrogen-enriched rings linked by C–C bond have the highest heats of formation spanning 996.11 kJ/mol (compound G), exceeding that of hexanitrohexaazaisowurtzitane (CL-20 = 397.8 kJ/mol)[34]. This indicates derivative G is more energetic but more unstable thermodynamically. However, kinetic stability is more important than thermal stability for high energy density molecules. Compared with nitroso compound (B), amino compound (A) has higher heats of formation. What's more, for these neutral compounds, when the -NHNO2 group is converted to -NHNH2, the heat of formation increases 164.06 kJ/mol from compound D to C. When the methoxy group is substituted by azido, the heat of formation for E increases to 996.11 kJ/mol, but unlike with other energetic groups, the introduction of -NNO2ONO2 lowers the heat formation. In general, comparing all derivatives, the presence of azido group gives rise to higher heats of formation than other groups. Thus we can conclude that -N3 is the superior group for increasing heat of formation, followed by the -NHNH2 and -NNO2NO2. The general trends of heats of formations of HOFs in all substituted derivatives are arranged as -N3 > -NHNH2 > -NNO2NO2 > -NH2 > -NNH2NO2 > -NHNO2 > -NO2 > -OCH3 > -ONO2 > -NNO2ONO2.
DownLoad:
CSV
| Compound | E0 (a.u.) | ZPE (a.u.) | HT (a.u.) | HOFg (kJ/mol) | HOFsub (kJ/mol) | HOFs (kJ/mol) |
| Parenta | 593.29 | |||||
| A | −1003.05796 | 0.13453 | 0.01625 | 395.26 | 69.05 | 326.21 |
| B | −1152.22866 | 0.11918 | 0.01741 | 289.11 | 60.90 | 228.20 |
| C | −1058.39313 | 0.15153 | 0.01738 | 471.16 | 68.14 | 403.02 |
| D | −1207.58976 | 0.13673 | 0.01840 | 297.99 | 59.06 | 238.93 |
| E | −1062.21543 | 0.14911 | 0.01760 | 264.36 | 61.56 | 202.80 |
| F | −1227.42263 | 0.12273 | 0.01859 | 198.12 | 60.26 | 137.86 |
| G | −1111.15299 | 0.11752 | 0.01779 | 1056.79 | 60.68 | 996.11 |
| H | −1262.92981 | 0.15268 | 0.01991 | 359.28 | 61.77 | 297.51 |
| I | −1412.01318 | 0.12795 | 0.02452 | 466.83 | 63.86 | 402.97 |
| J | −1487.28702 | 0.14069 | 0.02242 | 181.66 | 56.36 | 125.30 |
| RDXb | 95.14 | |||||
| TATBb | 102.67 | |||||
| aData from Ref. [19]. bData from Ref. [34]. |
||||||
Frontier molecular orbital theory has been widely used by chemists because molecular orbital analysis can provide valuable information for the study of their electronic structures[35, 36]. Thus, we paid a great deal of attention, not only to the highest occupied molecular orbitals (HOMOs) but also to the lowest unoccupied molecular orbitals (LUMOs). The gap energy (
As can be seen from Table 3, the HOMO energies vary from −0.3220 to −0.2436 a.u. and LUMO energies from −0.1269 to −0.1121 a.u. for all derivatives of the title compounds. It is proposed that smaller energy gap in the molecule is expected to be more reactive in the photochemical or chemical with electron leap or transfer. The electron withdrawing groups (-NO2) lower the LUMO and HOMO energy levels, whereas electron donating groups (-NH2, -NHNH2) can increase the LUMO and HOMO energy levels. When the electron withdrawing group is attached to the parent compound, it will reduce the stability of the substance. The electron donating group is the opposite. From the energy gap of all substituted compounds in Table 3, we can find that derivative A has the largest energy gap. That is to say, it is proposed that the molecule with the largest energy gap should have the lowest reactivity in chemical or photochemical processes involving an electron transfer or jump. This shows that compound A is more stable than others. Hence, in the explosive molecule design, incorporation with -NH2 is an effective means to decrease the sensitivity despite its disadvantage to detonation performance. However, it should be pointed out that the sensitivity is estimated using
DownLoad:
CSV
| Compound | HOMO | LUMO | H50 (cm) | |||
| Parent | −0.2419 | −0.1130 | 0.1290 | 232.83526 | 0.17215 | 32.0 |
| A | −0.3162 | −0.1136 | 0.2026 | 204.75801 | 0.21945 | 42.3 |
| B | −0.2904 | −0.1227 | 0.1677 | 242.92963 | 0.16029 | 29.4 |
| C | −0.2873 | −0.1121 | 0.1752 | 201.41270 | 0.21752 | 41.9 |
| D | −0.2565 | −0.1215 | 0.1350 | 249.94509 | 0.14764 | 26.6 |
| E | −0.3143 | −0.1127 | 0.2016 | 183.10390 | 0.19855 | 38.0 |
| F | −0.2546 | −0.1206 | 0.1340 | 248.91253 | 0.15411 | 28.0 |
| G | −0.2436 | −0.1269 | 0.1167 | 266.09794 | 0.14841 | 26.7 |
| H | −0.3220 | −0.1205 | 0.2015 | 228.78232 | 0.17175 | 31.9 |
| I | −0.2882 | −0.1254 | 0.1629 | 222.83197 | 0.18529 | 34.9 |
| J | −0.2620 | −0.1201 | 0.1420 | 241.95142 | 0.13754 | 24.5 |
| RDXc | 26.0 | |||||
| HMXc | 29.0 | |||||
| CL-20d | 12.0 | |||||
| cData from Ref. [37]. dData from Ref. [34]. |
||||||
In addition to the heat of formation and electronic properties, predicting the impact sensitivity of new organic molecules which are candidates to be energetic materials has been the topic of several studies in the past few years. The impact sensitivity can be evaluated by using characteristic height (H50). Pospíšil et al. have correlated the explosive characteristics of an energetic material to the electrostatic potential of the molecule and put forward an empirical formula relating H50 and electrostatic potential of the molecule, as given by equation (8). Table 4 shows a comparison of the impact sensitivity (H50) for derivatives with experimental and calculated H50 parameters for commonly used explosives. It can be seen from the table that all the explosive molecules exhibit relatively acceptable impact sensitivities with H50 values ranging from 24.5 to 42.3 cm, which means all derivatives surpass CL-20 (12.0 cm) and RDX (26.0 cm) except for new molecule J[33, 37]. Among them, compound A displays very high impact sensitivity (The H50 value is 42.3 cm). In contrast, A is much higher than HMX (32 cm)[37]. More importantly, derivatives A, C, E and I display lower sensitivity than the parent (H50 = 32 cm). This may suggest that properly replacing the hydrogen atom by high-energy group in explosives with super high energy may be a useful way to make them less sensitive without reducing too much energy.
DownLoad:
CSV
| Compound | Q(cal/g) | P(GPa) | D(km/s) | OB100 | N% | ||
| Parente | 1.88 | 36.20 | 9.07 | ||||
| A | 1.78 | 1.79 | 891.70 | 24.11 | 7.40 | −1.56 | 54.69 |
| B | 1.84 | 1.91 | 1563.04 | 36.05 | 8.95 | 0.70 | 48.95 |
| C | 1.69 | 1.72 | 878.41 | 22.03 | 7.18 | −1.85 | 56.83 |
| D | 1.83 | 1.83 | 1036.39 | 28.30 | 7.95 | 0.33 | 51.16 |
| E | 1.55 | 1.56 | 1126.10 | 19.60 | 6.97 | −2.06 | 40.33 |
| F | 1.85 | 1.85 | 1151.73 | 30.68 | 8.25 | 1.32 | 46.36 |
| G | 1.76 | 1.74 | 906.91 | 23.90 | 7.39 | −0.71 | 59.57 |
| H | 1.83 | 1.80 | 1610.09 | 37.31 | 9.12 | 0.00 | 53.16 |
| I | 1.82 | 1.78 | 1320.83 | 32.52 | 8.54 | 1.73 | 48.55 |
| J | 1.90 | 1.90 | 1218.66 | 34.39 | 8.66 | 2.21 | 46.41 |
| RDXf | 1.82 | 1345.57 | 39.00 | 9.10 | −21.61 | 37.84 | |
| HMXf | 1.91 | 1343.81 | 34.00 | 8.75 | −21.61 | 37.84 | |
| eData from Ref. [19]. fData from Ref. [38]. |
|||||||
Apart from sensitivity issues, the detonation performance of energetic materials is also of critical importance. Detonation velocity (D) and pressure (P) are elementary parameters for estimating the detonation performance of HEDMs. In order to assess the detonation velocities (D) and detonation pressures (P) of unsynthesized energetic complexes, we used the method based on K-J method equations (5) and (6), which has been widely used to predict the pressures and detonation velocities of many energetic complexes. The detonation velocity (D) and detonation pressure (P) are bound up with density (ρ) and heat of detonation (Q). Thus, Table 4 collected the predicted crystal densities (ρ), detonation velocities (D), detonation pressure (P) and heat of detonation (Q) of the title derivatives. For comparison, the ρ, D and P of the well-known explosives RDX and HMX are also listed in this table.
The densities of prepared compounds fall in the range of 1.56~1.91 g/cm3. We noticed that the actual densities of compound A may be higher, because -NH2 and -NO2 groups of adjacent molecules can be electrostatically attracted each other and pull the molecular units closer together in the condensed phase. Besides, the densities of molecules B, D, F, and J are higher than RDX (1.82 g/cm3)[38], meeting the standards for high-density-energy materials (HEDMs). As summarized in Table 4, compound B possesses the highest density of 1.91 g/cm3, which is over HMX (1.90 g/cm3)[38]. Similar to density, these new compounds exhibit high detonation heat (Q), and compounds B and H have the high values of 1563.04 and 1610.09 cal/g, indicating that the NH2 and NO2 groups can effectively increase the detonation heat of the system. By comparing physicochemical properties of the parent and derivatives (Table 4), we found that the introduction of one nitro to replace the hydrogen atom can greatly improve the density, which proved the validity of our original hypothesis. In our work, molecule B is inferior to H in view of its detonation heat (Q = 1563.04 cal/g), detonation velocity (D = 8.97 km/s) and detonation pressure (P = 36.05 GPa). It is ascribed to the less nitrogen content in compound B than in H. Thus, quite promisingly, with the increased nitrogen content, the molecules have much better D, P and Q values. At the same time, the detonation pressure and detonation velocity of molecule H are higher than that of the parent compound. The calculated D and P suggest that it has the potential to be a high-performance energetic material. Moreover, the cyclic organic molecule with polynitro functionality may be ideal energetic materials that can solve the long-standing inherent contradiction between detonation performance and stability.
Oxygen balance (OB) is another important index to evaluate the deficiency or excess of oxygen in a molecule required to convert all carbon into carbon dioxide and all hydrogen into water. As can be seen from Table 4, arising from the multiple nitro and nitramino functionalities, derivatives B, D, F, I and J have positive oxygen balances, whereas the OB value of H is zero. Other compounds exhibit negative OB values ranging from −2.06% to −0.71%. It is important to note that the OB value of compound H is zero, which indicates that it can fully use its chemical energy. Notably, the detonation performance (Q = 1610.09 cal/g, D = 9.12 km/s) of derivative H is much superior to the typically high explosives including RDX (Q = 1345.57 cal/g, D = 9.12 km/s)[38]. This is due to conjugate action and intramolecular hydrogen bonds in the system, and the high nitrogen content is also good for improving detonation performance. For energetic propellant fuels, high nitrogen content is advantageous for smokeless combustion. The nitrogen content of all derivatives ranged from 40.33% (compound E) to 59.57% (compound G), which is higher than those of RDX and HMX (N% = 37.84%). What's more, the excellent heat of formation of compound G is attributed to their high nitrogen content (N% = 59.57%), as well as the large number of high-energy bonds (C–N, N–N and N–O bonds), which is due to the unique structure. At the same time, the derivatives of title compounds are suggested to have potential for further study.
In this work, a series of novel nitrogen-enriched energetic derivatives with dicyclic structures have been designed. And the density, heat of formation, electronic properties, properties of detonation and impact sensitivity were calculated using the DFT method at the B3LYP/6-311G** level of the theory. In the design of new energetic materials, these compounds of nitrogen-rich heterocycles exhibit some favorable features:
(1) Our results showed that all derivatives possess positive HOFs. This changing trend of HOFs in substituted can be arranged in the sequence as -N3 > -NHNH2 > -NNO2NO2 > -NH2 > -NNH2NO2 > -NHNO2 > -NO2 > -OCH3 > -ONO2 > -NNO2ONO2. Quite noteworthy in this respect is molecule G with the most positive enthalpy of formation (996.11 kJ/mol) than that of other common energetic compounds and a large enthalpy of formation are very beneficial to the detonation heat.
(2) The triazole-based materials have relatively high stability due to the appropriate value of energy separation (
(3) The impact sensitivity (H50) of the new compounds fall in the range of 24.5 to 42.3 cm which is less sensitive than CL-20 (12.0 cm). The results indicate that these derivatives have preferable thermal stability. Aside from derivatives D, E, F and J, the H50 of the new organic molecules is higher than those for RDX and HMX, which implies that the sensitivities of the new compounds are lower than those of RDX and HMX. The stability of energetic materials is very momentous because they need long-term preservation and adaptation to various environments.
(4) The predicted results for these newly designed compounds exhibit good detonation performance (especially compounds B and H). Besides, compounds B and H (1.91 and 1.80 g/cm3) also show higher density than TATB (1.79 g/cm3)[39] and are comparable with HMX (1.91 g/cm3) and RDX (1.82 g/cm3)[38]. It is a remarkable fact that the nitrogen contents of all compounds have high nitrogen contents between 40.33% (molecule E) and 59.57% (molecule G). Compounds bearing high nitrogen content in compounds often cause energetic materials to exhibit good performance and show great potential as additives in gas generators, as insensitive ammunition, and as smoke-free pyrotechnics.
Based on the above results, we can conclude that organic compounds B (H50 = 29.4 cm, ρ = 1.91 g/cm3, Q = 1563.04 cal/g, P = 36.05 GPa, D = 8.95 km/s) and H (H50 = 31.9 cm, ρ = 1.80 g/cm3, Q = 1610.09 cal/g, P = 37.31 GPa, D = 9.12 km/s) can be considered a potential candidate for an HEDM. Our observations indicate that the combination of triazole derivatives and oxygen balance to zero is a very effective way to obtain potential energetic compounds with outstanding detonation performance. These compounds present good explosive potentials and are worthy of synthesis and further investigation. Our results should also provide some useful information for the molecular design of novel HEDCs.
Kumar, M. A.; Ashutosh, P.; Vargeese, A. A. Decomposition mechanism of hexanitrohexaazaisowurtzitane (CL-20) by coupled computational and experimental study. J. Phys. Chem. A 2019, 123, 4014–40203. doi: 10.1021/acs.jpca.9b01197
Zhang, J. C.; Zhu, Z. Y.; Zhou, M. Q.; Zhang J. H.; Hooper, J. P.; Shreeve, J. M. Superior high-energy-density biocidal agent achieved with a 3D metal-organic framework. ACS Appl. Mater. Interfaces 2020, 12, 40541−40547. doi: 10.1021/acsami.0c12251
Qu, R. J.; Liu, H. X.; Feng, M. B.; Yang, X.; Wang, Z. Y. Investigation on intramolecular hydrogen bond and some thermodynamic properties of polyhydroxylated anthraquinones. J. Chem. Eng. Data 2012, 57, 2442−2455. doi: 10.1021/je300407g
Park, A.; Jeong, Y.; Lee, T. K.; Park, M. W.; Lim, H. Y.; Sung, J.; Cho, J.; Kwak, S. K.; Hong, S. Y.; Choi, N. S. Replacing conventional battery electrolyte additives with dioxolone derivatives for high-energy-density lithium-ion batteries. Nat. Commun. 2021, 12, 838−850. doi: 10.1038/s41467-021-21106-6
Zhang, W. Q.; Zhang, J. H.; Deng, M. C.; Qi, X. J.; Nie, F. D.; Zhang, Q. H. A promising high-energy-density material. Nat. Commun. 2017, 8, 181−187. doi: 10.1038/s41467-017-00286-0
Liu, Z.; Lu, T.; Xue, F.; Nie, H. C.; Ray, W.; Andrew, S.; Felipe, K.; Narendirakumar, N.; Dong, X. L.; Yu, D. H.; Chen, L. Q.; Liu, Y.; Wang, G. S. Lead-free (Ag, K)NbO3 materials for high-performance explosive energy conversion. Sci Adv. 2020, 6, eaba0367−eaba0377. doi: 10.1126/sciadv.aba0367
Tang, Y. X.; He, C. L.; Imler, G. H.; Parrish, D. A.; Shreeve, J. M. Aminonitro groups surrounding a fused pyrazolotriazine ring: a superior thermally stable and insensitive energetic material. ACS Appl. Energy Mater. 2019, 2, 2263–2267. doi: 10.1021/acsaem.9b00049
Chang, J. J.; Zhao, G.; Zhao, X. Y.; He, C. L.; Pang, A. P.; Shreeve, J. M. New promises from an old friend: iodine-rich compounds as prospective energetic biocidal agents. Acc. Chem. Res. 2021, 54, 332–343. doi: 10.1021/acs.accounts.0c00623
Ma, J. C.; Zhang, J. H.; Imler, G. H.; Parrish, D. A.; Shreeve, J. M. Gem-dinitromethyl-functionalized 5-amino-1, 3, 4-oxadiazolate derivatives: alternate route, characterization, and property analysis. Org. Lett. 2020, 22, 4771–4775. doi: 10.1021/acs.orglett.0c01569
Ma, J. C.; Tang, Y. X.; Cheng, G. B.; Imler, G. H.; Parrish, D. A.; Shreeve, J. M. Energetic derivatives of 8-nitropyrazolo[1, 5-a][1, 3, 5]triazine-2, 4, 7-triamine: achieving balanced explosives by fusing pyrazole with triazine. Org. Lett. 2020, 22, 1321–1325. doi: 10.1021/acs.orglett.9b04642
Xu, H. J.; Peng, L. J.; Wang, J. B.; Ren, H. S.; Zhu, Q.; Li, X. Y. Relationship between energetic performance and clustering effects on incremental nitramine groups: a theoretical perspective. J. Phys. Chem. A 2019, 123, 742–749. doi: 10.1021/acs.jpca.8b10647
Talawar, M. B.; Sivabalan, R.; Mukundan, T.; Muthurajan, H.; Sikder, A. K.; Gandhe, B. R.; Rao, A. S. Environmentally compatible next generation green energetic materials (GEMs). J. Hazard. Mater. 2009, 161, 589–607. doi: 10.1016/j.jhazmat.2008.04.011
Witkowski, T. G.; Sebastiao, E.; Gabidullin, B.; Hu, A.; Zhang, F.; Murugesu, M. 2, 3, 5, 6-tetra(1H-tetrazol-5-yl)pyrazine: a thermally stable nitrogen-rich energetic material. ACS Appl. Energy Mater. 2018, 1, 589–593. doi: 10.1021/acsaem.7b00138
Zhai, L. J.; Bi, F. Q.; Zhang, J. R.; Li, X. Z.; Wang, B. Z.; Chen, S. P. 3, 4-Bis(3-tetrazolylfuroxan-4-yl)furoxan: a linear C–C bonded pentaheterocyclic energetic material with high heat of formation and superior performance. ACS Omega 2020, 5, 11115–11122. doi: 10.1021/acsomega.0c01048
Dong, Z.; Ye, Z. W. Synthesis and properties of salts derived from C4N182-, C4N18H3- and C4N18H3- anions. J. Mater. Chem. A 2020, 8, 25035–25039. doi: 10.1039/D0TA08153A
Bagdi, P. R.; Basha, R. S.; Baruah, P. K.; Khan, A. T. Copper oxide nanoparticle mediated 'click chemistry' for the synthesis of mono-, bis- and tris-triazole derivatives from 10, 10-dipropargyl-9-anthrone as a key building block. RSC Adv. 2014, 4, 10652–10659. doi: 10.1039/c3ra44869j
Nahle, A.; Salim, R.; Hajjaji, F. E.; Aouad, M. R.; Messali, M.; Ech-chihbi, E.; Hammouti, B.; Taleb, M. Novel triazole derivatives as ecological corrosion inhibitors for mild steel in 1.0 M HCl: experimental & theoretical approach. RSC Adv. 2021, 11, 4147–4162. doi: 10.1039/D0RA09679B
Sharma, J.; Ahmad, S.; Alam, M. S. Bioactive triazoles: a potential review. J. Chem. Pharm. Res. 2012, 4, 5157−5164.
Xu, Z.; Cheng, G. B.; Zhu, S. G.; Lin, Q. H.; Yang, H. W. Nitrogen-rich salts based on the combination of 1, 2, 4-triazole and 1, 2, 3-triazole rings: a facile strategy for fine tuning energetic properties. J. Mater. Chem. A 2018, 6, 2239–2248. doi: 10.1039/C7TA08941D
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B. G.; Petersson, A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery Jr., J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J.; Fox, D. J. Gaussian 09, Revision D. 01, Gaussian, Inc., Wallingford CT 2009.
Shi, J. Q.; Qu, R. J.; Feng, M. B.; Wang, X. H.; Wang, L. S.; Yang, S. G.; Wang, Z. Y. Oxidative degradation of decabromodiphenyl ether (BDE 209) by potassium permanganate: reaction pathways, kinetics, and mechanisms assisted by density functional theory calculations. Environ. Sci. Technol. 2015, 49, 4209–4217. doi: 10.1021/es505111r
Qu, R. J.; Xu, B. Z.; Meng, L. J.; Wang, L. S.; Wang, Z. Y. Ozonation of indigo enhanced by carboxylated carbon nanotubes: performance optimization, degradation products, catalytic mechanism and toxicity evaluation. Water Res. 2015, 68, 316–327. doi: 10.1016/j.watres.2014.10.017
Hehre, W. J.; Ditchfield, D.; Radom, L.; Pople, J. A. Molecular orbital theory of the electronic structure of organic compounds. V. Molecular theory of bond separation. J. Am. Chem. Soc. 1970, 92, 4796–4801. doi: 10.1021/ja00719a006
Zhang, J. C.; Zhang, J. H.; Imler, G. H.; Parrish, D. A.; Shreeve, J. M. Sodium and potassium 3, 5-dinitro-4-hydropyrazolate: three dimensional metal-organic frameworks as promising super-heatresistant explosives. ACS Appl. Energy Mater. 2019, 2, 7628–7634. doi: 10.1021/acsaem.9b01608
Zhao, G.; He, C. L.; Yin, P.; Imler, G. H.; Parrish, D. A.; Shreeve, J. M. Efficient construction of energetic materials via nonmetallic catalytic carbon-carbon cleavage/oxime-release-coupling reactions. J. Am. Chem. Soc. 2018, 140, 3560–3563. doi: 10.1021/jacs.8b01260
He, P.; Zhang, J. G.; Wang, K.; Yin, X.; Jin, X.; Zhang, T. L. Extensive theoretical studies on two new members of the FOX-7 family: 5-(dinitromethylene)-1, 4-dinitramino-tetrazole and 1, 1΄-dinitro-4, 4΄-diamino-5, 5΄-bitetrazole as energetic compounds. Phys. Chem. Chem. Phys. 2015, 17, 5840–5848. doi: 10.1039/C4CP04883K
Jin, X. H.; Hu, B. C.; Lu, W.; Gao, S. J.; Liu, Z. L.; Lv, C. X. Theoretical study on a novel high-energy density material 4, 6, 10, 12-tetranitro-5, 11-bis(nitroimino)-2, 8-dioxa-4, 6, 10, 12 -tetraaza-tricyclo[7, 3, 0, 03, 7] dodecane. RSC Adv. 2014, 4, 6471–6477. doi: 10.1039/c3ra46107f
Politzer, P.; Lane, P.; Murray, J. S. Computational characterization of a potential energetic compound: 1, 3, 5, 7-tetranitro-2, 4, 6, 8-tetraazacubane. Cent. Eur. J. Energ. Mater. 2011, 8, 39–52.
Rice, B. M.; Pai, S. V.; Hare, J. Predicting heats of formation of energetic materials using quantum mechanical calculations. Combust. Flame. 1999, 118, 445–458. doi: 10.1016/S0010-2180(99)00008-5
Kamlet, M. J.; Jacobs, S. J. Chemistry of detonations. I. A simple method for calculating detonation properties of C–H–N–O explosives. J. Chem. Phys. 1968, 48, 23–35. doi: 10.1063/1.1667908
Politzer, P.; Murray, J. S. The fundamental nature and role of the electrostatic potential in atoms and molecules. Theor. Chem. Acc. 2002, 108, 134–142. doi: 10.1007/s00214-002-0363-9
Pospíšil, M.; Vávra, P.; Concha, M. C.; Murray, J. S.; Politzer, P. A possible crystal volume factor in the impact sensitivities of some energetic compounds. J. Mol. Model. 2010, 16, 895–901. doi: 10.1007/s00894-009-0587-x
Zhang, C.; Shu, Y.; Huang, Y.; Zhao, X.; Dong, H. Investigation of correlation between impact sensitivities and nitro group charges in nitro compounds. J. Phys. Chem. B 2005, 109, 8978–8982. doi: 10.1021/jp0512309
Li, B. T.; Li, L. L.; He, J. X. Looking for high energy density molecules in the nitro-substituted derivatives of pyridazine. Chin. J. Struct. Chem. 2020, 39, 849–854.
Li, Y.; Evans, J. N. S. The Fukui function: a key concept linking frontier molecular orbital theory and the hard-soft-acid-base principle. J. Am. Chem. Soc. 1995, 117, 7756–7759. doi: 10.1021/ja00134a021
Ess, D. H.; Houk, K. N. Theory of 1, 3-dipolar cycloadditions: distortion/interaction and frontier molecular orbital models. J. Am. Chem. Soc. 2008, 130, 10187–10198. doi: 10.1021/ja800009z
Guo, C.; Zhang, H.; Wang, X.; Liu, X.; Sun, J. Study on a novel energetic cocrystal of TNT/TNB. J. Mater. Sci. 2013, 48, 1351–135. doi: 10.1007/s10853-012-6881-5
Gutowski, K. E.; Rogers, R. D.; Dixon, D. A. Accurate thermochemical properties for energetic materials applications. II. Heats of formation of imidazolium-, 1, 2, 4-triazolium-, and tetrazolium-based energetic salts from isodesmic and lattice energy calculations. J. Phys. Chem. B 2007, 109, 4788–4800.
Boddu, V. M.; Viswanath, D. S.; Ghosh, T. K.; Damavarapu, R. 2, 4, 6-Triamino-1, 3, 5-trinitrobenzene (TATB) and TATB-based formulations - a review. J. Hazard. Mater. 2010, 181, 1−8. doi: 10.1016/j.jhazmat.2010.04.120

Figure 1 Derivatives of 4-nitro-5-(5-nitroimino-1, 2, 4-triazol-3-yl)-2H-1, 2, 3-triazolate designed in this paper
Table 1.
Calculated Methods for the Values of N,
| Parameters | Explosives components conditions | ||
| c≥2a+b/2 | 2a+b/2 > c≥b/2 | b/2 > c | |
| (b+2c+2d)/4M | (b+2c+2d)/4M | (b+d)/2M | |
| 4M/(b+2c+2d) | (56d+88c-8b)/(b+2c+2d) | (2b+28d+32c)/(b+d) | |
| Q*10-3 | (28.9b+94.05a +0.239 |
[28.9b+94.05(c/2-b/4) +0.239 |
(57.8c+0.239 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Total Energies (E0), Zero Point Energies (ZPE), Thermal Correction Values (HT), and Heat of Formation (HOF) of All the Derivatives at the B3LYP/6-311G** Level of Theory Compared to RDX and TATB
| Compound | E0 (a.u.) | ZPE (a.u.) | HT (a.u.) | HOFg (kJ/mol) | HOFsub (kJ/mol) | HOFs (kJ/mol) |
| Parenta | 593.29 | |||||
| A | −1003.05796 | 0.13453 | 0.01625 | 395.26 | 69.05 | 326.21 |
| B | −1152.22866 | 0.11918 | 0.01741 | 289.11 | 60.90 | 228.20 |
| C | −1058.39313 | 0.15153 | 0.01738 | 471.16 | 68.14 | 403.02 |
| D | −1207.58976 | 0.13673 | 0.01840 | 297.99 | 59.06 | 238.93 |
| E | −1062.21543 | 0.14911 | 0.01760 | 264.36 | 61.56 | 202.80 |
| F | −1227.42263 | 0.12273 | 0.01859 | 198.12 | 60.26 | 137.86 |
| G | −1111.15299 | 0.11752 | 0.01779 | 1056.79 | 60.68 | 996.11 |
| H | −1262.92981 | 0.15268 | 0.01991 | 359.28 | 61.77 | 297.51 |
| I | −1412.01318 | 0.12795 | 0.02452 | 466.83 | 63.86 | 402.97 |
| J | −1487.28702 | 0.14069 | 0.02242 | 181.66 | 56.36 | 125.30 |
| RDXb | 95.14 | |||||
| TATBb | 102.67 | |||||
| aData from Ref. [19]. bData from Ref. [34]. |
||||||
下载: 导出CSV
Table 3.
Energies of the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO), and Energy Gaps (
| Compound | HOMO | LUMO | H50 (cm) | |||
| Parent | −0.2419 | −0.1130 | 0.1290 | 232.83526 | 0.17215 | 32.0 |
| A | −0.3162 | −0.1136 | 0.2026 | 204.75801 | 0.21945 | 42.3 |
| B | −0.2904 | −0.1227 | 0.1677 | 242.92963 | 0.16029 | 29.4 |
| C | −0.2873 | −0.1121 | 0.1752 | 201.41270 | 0.21752 | 41.9 |
| D | −0.2565 | −0.1215 | 0.1350 | 249.94509 | 0.14764 | 26.6 |
| E | −0.3143 | −0.1127 | 0.2016 | 183.10390 | 0.19855 | 38.0 |
| F | −0.2546 | −0.1206 | 0.1340 | 248.91253 | 0.15411 | 28.0 |
| G | −0.2436 | −0.1269 | 0.1167 | 266.09794 | 0.14841 | 26.7 |
| H | −0.3220 | −0.1205 | 0.2015 | 228.78232 | 0.17175 | 31.9 |
| I | −0.2882 | −0.1254 | 0.1629 | 222.83197 | 0.18529 | 34.9 |
| J | −0.2620 | −0.1201 | 0.1420 | 241.95142 | 0.13754 | 24.5 |
| RDXc | 26.0 | |||||
| HMXc | 29.0 | |||||
| CL-20d | 12.0 | |||||
| cData from Ref. [37]. dData from Ref. [34]. |
||||||
下载: 导出CSV
Table 4.
Predicted Density, Explosive Heats, Detonation Pressure and Detonation Velocity of All Derivatives together with RDX and HMX (
| Compound | Q(cal/g) | P(GPa) | D(km/s) | OB100 | N% | ||
| Parente | 1.88 | 36.20 | 9.07 | ||||
| A | 1.78 | 1.79 | 891.70 | 24.11 | 7.40 | −1.56 | 54.69 |
| B | 1.84 | 1.91 | 1563.04 | 36.05 | 8.95 | 0.70 | 48.95 |
| C | 1.69 | 1.72 | 878.41 | 22.03 | 7.18 | −1.85 | 56.83 |
| D | 1.83 | 1.83 | 1036.39 | 28.30 | 7.95 | 0.33 | 51.16 |
| E | 1.55 | 1.56 | 1126.10 | 19.60 | 6.97 | −2.06 | 40.33 |
| F | 1.85 | 1.85 | 1151.73 | 30.68 | 8.25 | 1.32 | 46.36 |
| G | 1.76 | 1.74 | 906.91 | 23.90 | 7.39 | −0.71 | 59.57 |
| H | 1.83 | 1.80 | 1610.09 | 37.31 | 9.12 | 0.00 | 53.16 |
| I | 1.82 | 1.78 | 1320.83 | 32.52 | 8.54 | 1.73 | 48.55 |
| J | 1.90 | 1.90 | 1218.66 | 34.39 | 8.66 | 2.21 | 46.41 |
| RDXf | 1.82 | 1345.57 | 39.00 | 9.10 | −21.61 | 37.84 | |
| HMXf | 1.91 | 1343.81 | 34.00 | 8.75 | −21.61 | 37.84 | |
| eData from Ref. [19]. fData from Ref. [38]. |
|||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: