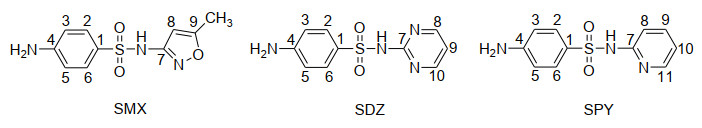
Figure 1.
Molecular structures of sulfamethoxazole (SMX), sulfadiazine (SDZ) and sulfapyridine (SPY) and the numbering system of C atoms

Sulfonamides (SAs) are one of the most widely used antibiotics and have been used for more than 50 years, mainly for livestock and aquaculture processes, and also for the treatment of human diseases because of their excellent broad-spectrum antibacterial properties and low prices[1]. SAs can enter the aquatic environment through the discharge of production and application processes, and have become a new type of environmental pollutants[2]. Currently, they have been widely detected in various environmental water bodies such as municipal sewage, domestic sewage, surface water and ground- water[3-8]. SAs persist in the water environment and present a "false" persistence phenomenon due to their continuous production, usage and discharge. The related studies have shown that residual SAs in water can lead to the production of drug-resistant bacteria and resistance genes, increase the risk of resistance to pathogenic bacteria, and show signi- ficant ecotoxicity[9-11]. SAs antibiotic pollution in water poses a serious threat to the ecological environment and human health, and has attracted the attention of the public and researchers. However, SA antibiotics cannot be effectively removed using con- ventional municipal sewage water treatment pro- cesses[12-14].
The advanced oxidation processes (AOPs) inclu- ding photocatalytic degradation are considered to be an advanced treatment technology that effectively removes trace organic contaminants from water[15, 16]. They can generate strong oxidizing substances such as OH radicals in situ, thereby degrading and even mineralizing most refractory organic pollutants. Compared with traditional methods, AOPs have received more and more attention due to their high efficiency in removing and degrading pollutants and being environmentally friendly. For example, OH radical was found to play a key role in the photocatalytic degradation[17-21] or other AOPs[22, 23] of SAs. Obviously, the initial reactions of organic pollutants with OH radicals would have an important influence on the ultimate degradation efficiency of organic pollutants in the advanced oxidation pro- cesses involving OH radical.
An important feature of SA molecules is that they all contain a benzene ring and a heterocyclic ring, which can both be attacked by OH radical. Another key characteristic is that they can exist in three different forms, namely cationic, neutral and anionic forms in aqueous solution due to simultaneous presence of basic amine group (-NH2) and acidic amide group (-NH-), which form dominates depen- dent on the solution pH. This makes the degradation of SA compounds involving OH radicals in aqueous solutions more complicated. Very recent experimen- tal observation indicated that OH reaction of SAs can experience multi-site hydroxylation and OH oxida- tion kinetics varied depending on the dominant form of SAs as a function of pH[24].
Quantum chemical calculation has an advantage in revealing reaction mechanisms and predicting degradation products of organic pollutants and has become one of the important means to study the environmental transformation process of organic pollutants. It was successfully applied to investigate photodegradation mechanism and products of several SAs[25-27], OH-initiated atmospheric oxidation degra- dation of some persistent organic pollutants (POPs)[28-32] and some other processes[33-36]. However, no comprehensive researches involving OH reactions of three different existing forms of SAs have been reported until now. In this paper, we conducted a comprehensive computational investigation on OH addition reactions of cationic, neutral and anionic forms of three selected SAs (sulfamethoxazole, sulfadiazine and sulfapyridine) in aqueous solution employing density functional theory (DFT) method. The aim of the present study is to illustrate the mechanism and potential energy surface of the investigated OH addition reactions, explore the OH addition reactivity of three different existing forms of the selected SAs and determine the most favorable OH addition position. The obtained results will help to understand environmental transformation pro- cesses of SAs involving OH radicals in real aquatic environment.
Prior researches[37-39] indicated that M06-2X method[40], a hybrid DFT method, is applicable to study molecular properties and reaction mechanism. In this work, M06-2X method and 6-311+G(3df, 2p) basis set were combined to optimize the molecular structures of reactants, products and transition states included in the studied reactions. On the basis of optimized structures of these stationary points, the vibrational frequency calculation was performed to ascertain their characteristics and to obtain their thermodynamic data such as Gibbs free energy. The solvent effect was considered by performing self-consistent reaction field (SCRF) optimization calculation using polarizable continuum model (PCM) method and employing water as a solvent[41, 42]. All calculations were carried out by using the Gaussian 09 program[43].
Traditional transition state theory (TST)[44] was utilized to compute the rate coefficients of OH addition reactions involved in this work. Eq. (1) was adopted for bimolecular elementary reactions (k, in M−1⋅s−1).
|
|
(1) |
where the meanings of all involved parameters were elucidated in our earlier study[30, 31].
Previous studies[45, 46] have shown that there are two main reactions between organic compounds and OH radicals in aqueous solution, namely direct hydrogen abstraction reactions and OH addition reactions on the aromatic ring. Moreover, OH addition to aromatic ring was found to be much faster than the corresponding H abstraction[45, 47]. Therefore, only OH addition reactions of three selected SAs were involved in this work. For ease discussion, the cationic, neutral, and anionic forms of three selected SAs are represented by SA+, SA, and SA−, respec- tively.
Fig. 1 denotes the molecular structures of neutral form of three SAs as well as their C atomic numbering. Due to C1 symmetry of these SAs, their OH addition reactions can occur at nine, ten or eleven different C positions, respectively.
It was found that OH addition reactions of cationic, neutral and anionic forms of sulfamethoxazole (SMX+, SMX and SMX−) are all bimolecular elementary reactions. When OH radicals and SMX (SMX+ or SMX−) approach with each other, SMX (SMX+ or SMX−)-OH adducts can be obtained through corresponding transition states (TSs). The forming C−O distances in TSs range over 1.97~2.25 Å, while these distances for the corresponding adducts fall in the range of 1.38~1.44 Å. An interesting finding is that the forming C−O distance in TSs related to SMX is always longer than that in the TSs involved in SMX+ at the same position, while the distance involving SMX− is also always longer than the corresponding one involving SMX.
Fig. 2 indicates nine OH addition reactions of SMX along with Gibbs free energies (Gr) of TSs and nine adducts relative to SMX + OH. Fig. S1 displays Gibbs free energy profiles for those of SMX+ and SMX−. It should be noted that Gr of TSs and adducts are Gibbs free energy of activation (ΔrG≠) and Gibbs free energy of reaction (ΔrG) of addition reactions, respectively. It can be seen from Fig. 2 that the differences of ΔrG (ΔrG≠) in gas phase and aqueous solution are all lower than 3 kcal⋅mol−1, demon- strating that the effect of water solvent on the free energy potential profile of the addition reaction of SMX neutral molecules is small. On the other hand, the data in Fig. S1 indicate that in most cases of OH addition reactions of SMX+ and SMX−, the ΔrG (ΔrG≠) values in aqueous solution are obviously higher than those in gas phase, which means that water solvent disfavors the OH addition reactions of SMX+ and SMX−. The probable cause is that the stabilizing effect of water solvent on the transition states and adducts of these addition reactions is not as strong as that on SMX+ or SMX−. Unless otherwise specified, the following discussion is based primarily on the results in aqueous solution.
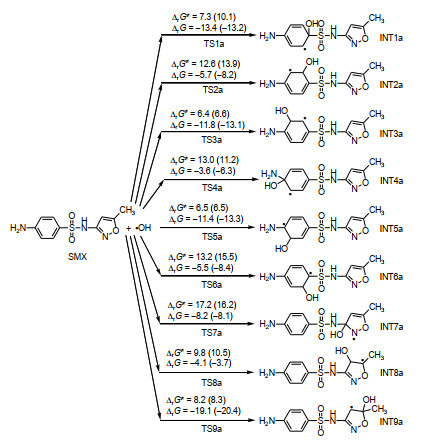
According to Fig. 2 and Fig. S1, all addition reactions have negative ΔrG values, suggesting that OH additions of SMX (SMX+ or SMX−) are thermodynamically spontaneous processes in the standard states. The ΔrG values for OH addition reactions of SMX+, SMX and SMX− are in the scopes of −3.8~−19.1, −3.8~−19.1 and −2.8~−20.1 kcal⋅mol−1, respectively. Although the adduct with addition occurring on the C(9) atom of SMX+, SMX or SMX− is the thermodynamically most stable in all three cases, the relative thermodynamic stability sequence of the other adducts is not uniform in the three cases.
However, the distribution of SMX (SMX+ or SMX−)-OH adducts was not decided by thermody- namic factors. Since all addition reactions are thermodynamic spontaneous processes, the distribu- tion of addition products is determined by the kinetic factors, i.e., the relative magnitude of ΔrG≠ of each reaction. According to Fig. 2 and Fig. S1, the ΔrG≠ values for OH addition reactions of SMX, SMX+ and SMX− are in the scopes of 10.3~17.8, 6.4~17.2 and 6.0~14.8 kcal⋅mol−1, respectively. Moreover, the addition reaction at the C(7) position always has the highest ΔrG≠ value in all three cases, suggesting that it is the most unfavorable kinetically. However, the most kinetically favorable reaction is not the same in three cases. Specifically, the kinetically most favorable reactions in the three cases are the additions to C(9), C(3), and C(5) sites, respectively.
The most striking feature related to OH addition reactions of SMX, SMX+ or SMX− is that for addi- tion reaction occurring at the same carbon position the ΔrG≠ value associated with SMX+ is always greater than the corresponding value for SMX, while the ΔrG≠ value associated with SMX− is always less than the corresponding value involving SMX. In other words, SMX− has the highest OH addition reactivity in three forms of SMX, while SMX+ is the least active, which is in good agreement with the recent experimental observation[24]. This result can be rationalized in terms of frontier molecular orbital (FMO) theory. The transfer of charge from SMX+, SMX or SMX− to OH was observed in the transition states of three corresponding OH addition reactions, indicating that the governing FMO interactions contained in these OH addition reactions are those between HOMO of SMX+, SMX or SMX− and LUMO of OH radicals and that these OH addition reactions are typical electrophilic addition reactions. Table 1 lists the calculated FMO energies of SMX+, SMX and SMX−, from which one can see that SMX− has the highest HOMO energy and SMX+ has the lowest HOMO energy. Therefore, SMX− should be the most reactive in OH addition reaction, while SMX+ has the lowest addition activity. Furthermore, it can also be found that three largest differences of ΔrG≠ value between addition reactions of SMX and SMX+ (7.8, 7.2 and 5.6 kcal⋅mol−1) are those involving addition occurring at C(1), C(5) and C(3) positions, while additions at C(6), C(4) and C(8) positions correspond to three largest differences of ΔrG≠ value between addition reactions of SMX and SMX− (4.4, 3.6 and 2.8 kcal⋅mol−1). These results are apparently due to differences in the electronic structures of SMX+, SMX and SMX−, including FMO energies and atomic charge distribution in molecules.
 DownLoad:
CSV
DownLoad:
CSV
| Sulfonamides | HOMO/a.u. | LUMO/a.u. |
| SMX+ | −0.32880 | −0.04519 |
| SMX | −0.27947 | −0.00930 |
| SMX− | −0.26225 | 0.00467 |
| SDZ+ | −0.32378 | −0.04379 |
| SDZ | −0.27771 | −0.02385 |
| SDZ− | −0.25920 | −0.00126 |
| SPY+ | −0.31495 | −0.04348 |
| SPY | −0.27878 | −0.01488 |
| SPY− | −0.25403 | 0.00465 |
Eq. (1) was utilized to estimate the rate constants for all possible OH addition reactions of SMX+, SMX and SMX− at 298.15 K (kbi, 298.15 K) and Table S1 lists these results. Using the expression Ri = ki/∑ki, in which ki denotes the rate constant of the ith reaction, the branching ratio (Ri) of each adduct can be computed based on assumption that the OH addition of SMX+, SMX and SMX− is irreversible. Table S1 presents all R values. As can be seen from Table S1, the overall R value for OH addition occurring at three C-sites of SMX oxazole ring is only 0.024, revealing that OH addition reactions of SMX mainly occur on the benzene ring. Furthermore, R for OH addition occurring at C(2), C(4) and C(6)-sites is all actually zero, suggesting that only C(1), C(3) and C(5)-positions on the benzene ring are significant for the OH addition reactions of SMX. On the other hand, although OH addition reactions of SMX− also occur primarily on the benzene ring, the addition at C(5)-site dominates. However, the situation for OH addition reactions of SMX+ is completely different. The total R value for OH addition taking place at six C-sites of SMX+ benzene ring is only 0.061, indicating that OH addition reactions of SMX+ mainly occur on the oxazole ring. Moreover, C(9)-site on the oxazole ring of SMX+ is far more advantageous than the other two positions for OH addition. These results can also be explained according to FMO theory. As stated above, the main FMO interactions in OH addition reactions of SMX+, SMX or SMX− are those between HOMO of SMX+, SMX or SMX− and LUMO of OH radicals. It can be found that in HOMO of SMX+, SMX or SMX− (bonding π molecular orbital), C(9), C(3) or C(5) atom has the largest 2p atomic orbital coefficient, respectively. Therefore, the OH addition reaction occurring on C(9), C(3) or C(5) atom of SMX+, SMX or SMX− is kinetically the most advantageous. The overall rate constants for OH addition reactions of SMX+, SMX and SMX− at 298.15 K (kbi, 298.15 K) are listed in Table 2, in which the calculated value for SMX is 6.93 × 109 M–1⋅s–1. This result agrees well with the previous experimental value (8.5 × 109 M–1⋅s–1)[48], indicating that the method applied in this work is suitable.
To compare OH addition reactivity of different sulfonamides and probe the effect of heterocycles in sulfonamides (for example, oxazole ring in sulfa- methoxazole), OH addition reactions of sulfadiazine (SDZ) and sulfapyridine (SPY) in aqueous solution were also investigated using the method described above (the molecular structures of SDZ and SPY are shown in Fig. 1). The obtained results showed that OH addition reactions of three forms of SDZ and SPY are also bimolecular elementary reactions. Gibbs free energy profiles for OH addition of three forms of SDZ and SPY are illustrated in Fig. S1. The data related to SDZ (SPY) and their OH addition reactions such as FMO energies, rate constants and R values of adduct are listed in Table 1, Table 2 or Table S1, respectively.
DownLoad:
CSV
| Reactions | k/M–1⋅s–1 |
| SMX+ + OH → SMX+-OH adducts | 4.75 × 106 |
| SMX + OH → SMX-OH adducts | 6.93 × 109 |
| SMX− + OH → SMX−-OH adducts | 1.42 × 1011 |
| SDZ+ + OH → SDZ+-OH adducts | 1.52 × 106 |
| SDZ + OH → SDZ-OH adducts | 2.00 × 1010 |
| SDZ− + OH → SDZ−-OH adducts | 3.93 × 1010 |
| SPY+ + OH → SPY+-OH adducts | 8.85 × 106 |
| SPY + OH → SPY-OH adducts | 1.99 × 109 |
| SPY− + OH → SPY−-OH adducts | 4.22 × 1011 |
As can be seen from Table 2, the OH addition reactions of three different SAs have a common feature, that is, their anionic form is more reactive than the neutral molecule, while the neutral molecule is more active than the corresponding cation. Obviously, this is because, regardless of which SA, its anionic form always has the highest HOMO energy, and the cationic form always has the lowest HOMO energy. However, there is some degree of difference in their OH addition reactions. For the OH addition reaction of SMX and SDZ, the difference in the rate constant using the neutral molecule or the cation as an addition reagent, respectively is much larger than that in the rate constant involving the anion and the neutral molecule. For example, the total rate constant (k) of the OH addition reaction of SMX is about 1460 times the corresponding k value for SMX+, while the k value for SMX− is only 20 times the k value for SMX. In the other hand, for the OH addition reaction of SPY, the ratio of two k values for SPY and SPY+ (220) is almost the same as that involving SPY and SPY− (210). This result can also be illustrated using FMO theory. It can be derived from Table 1 that the difference of HOMO energy between SMX and SMX+ is obviously larger than that between SMX and SMX−, and the same is true for HOMO energy of SDZ, SDZ+ and SDZ−. That is, for SMX and SDZ, the difference in HOMO level between cations and neutral molecules is obviously greater than that between anions and neutral molecules. However, the difference in HOMO energy between SPY and SPY− is close to that between SPY and SPY+. Therefore, the situation in the OH addition reactions of SPY is not different from that involving SMX or SDZ to some extent.
We can see from Table S1 that OH addition reactions of SDZ and SDZ− mainly occur on the benzene ring, while both benzene and pyrimidine rings are important for OH addition reactions of SDZ+. In addition, the position selectivity for OH addition reactions of SDZ is different from that involving SDZ−. Addition at C(3)-site dominates in the reactions of SDZ, while both C(3) and C(5)-positions on the benzene ring are important for OH addition reactions of SDZ−. As for OH addition reactions of three forms of SPY, the situation involving neutral SPY is similar to that relating to neutral SMX or SDZ, i.e., OH addition reactions of neutral SPY also mainly occur on the benzene ring. However, the OH addition reaction involving cationic or anionic SPY is not the same as the corresponding reaction using cationic or anionic SMX (SDZ) as addition reagent. Specifically, the OH addition reaction of SPY+ mainly occurs on the benzene ring, while both the benzene and the pyridine rings are important for the OH addition reaction of SPY−. In addition, it was also found that for the OH addition reactions mainly occurring on the benzene ring, C(3) or (and) C(5) atoms are always the most favorable addition positions.
A systematic computational investigation of OH addition reactions of neutral, cationic and anionic forms of three sulfonamides in aqueous solution was performed using M06-2X/6-311+G(3df, 2p) method in the present study. The following are some important conclusions that have been obtained. OH addition reactions of sulfonamides are all thermo- dynamically spontaneous processes at standard conditions. The reactivity order of OH addition reaction of the three forms of sulfonamides in aqueous solution from high to low is: anion > neutral molecule > cation, which can be illustrated using FMO theory. OH addition reactions of neutral forms of three sulfonamides mainly occur in benzene ring. The addition reaction occurring on the benzene ring mainly involves C(3) or (and) C(5) atoms. Water solvent disfavors OH addition reactions of cationic and anionic forms of sulfonamides, however, it has only an insignificant effect on OH addition reactions of neutral sulfonamides.
Managaki, S.; Murata, A.; Takada, H.; Tuyen, B. C.; Chiem, N. H. Distribution of macrolides, sulfonamides, and trimethoprim in tropical waters: ubiquitous occurrence of veterinary antibiotics in the Mekong Delta. Environ. Sci. Technol. 2007, 41, 8004−8010. doi: 10.1021/es0709021
Luo, Y.; Xu, L. Q.; Rysz, M.; Wang, Y.; Zhang, H.; Alvarez, P. J. J. Occurrence and transport of tetracycline, sulfonamide, quinolone, and macrolide antibiotics in the Haihe River Basin. Environ. Sci. Technol. 2011, 45, 1827−1833. doi: 10.1021/es104009s
Baran, W.; Adamek, E.; Ziemianska, J.; Sobczak, A. Effects of the presence of sulfonamides in the environment and their influence on human health. J. Hazard. Mater. 2011, 196, 1−15. doi: 10.1016/j.jhazmat.2011.08.082
Zhang, R. J.; Zhang, G.; Zheng, Q.; Tang, J. H.; Chen, Y. J.; Xu, W. H.; Zou, Y. D.; Chen, X. X. Occurrence and risks of antibiotics in the Laizhou Bay, China: impacts of river discharge. Ecotox. Environ. Safe. 2012, 80, 208−215. doi: 10.1016/j.ecoenv.2012.03.002
Shimizu, A.; Takada, H.; Koike, T.; Takeshita, A.; Saha, M.; Nakada, N.; Murata, A.; Suzuki, T.; Suzuki, S.; Chiem, N. H.; Tuyen, B. C.; Viet, P. H.; Siringan, M. A; Kwan, C.; Zakaria, M. P.; Reungsang, A. Ubiquitous occurrence of sulfonamides in tropical Asian waters. Sci. Total Environ. 2013, 452−453, 108−115.
Bahnmüller, S.; Gunten, U. V.; Canonica, S. Sunlight-induced transformation of sulfadiazine and sulfamethoxazole in surface waters and wastewater effluents. Water Res. 2014, 57, 183−192. doi: 10.1016/j.watres.2014.03.019
Ma, Y.; Li, M.; Wu, M.; Li, Z.; Liu, X. Occurrences and regional distributions of 20 antibiotics in water bodies during groundwater recharge. Sci. Total Environ. 2015, 518, 498−506.
Yao, L.; Wang, Y.; Tong, L.; Deng, Y.; Li, Y.; Gan, Y.; Guo, W.; Dong, C.; Duan, Y.; Zhao, K. Occurrence and risk assessment of antibiotics in surface water and groundwater from different depths of aquifers: a case study at Jianghan Plain, central China. Ecotox. Environ. Safe. 2017, 135, 236−242. doi: 10.1016/j.ecoenv.2016.10.006
Baran, W.; Sochacka, J.; Wardas, W. Toxicity and biodegradability of sulfonamides and products of their photocatalytic degradation in aqueous solutions. Chemosphere 2006, 65, 1295−1299. doi: 10.1016/j.chemosphere.2006.04.040
Huang, D. J.; Hou, J. H.; Kuo, T. F.; Lai, H. T. Toxicity of the veterinary sulfonamide antibiotic sulfamonomethoxine to five aquatic organisms. Environ. Toxicol. Pharmacol. 2014, 38, 874−880. doi: 10.1016/j.etap.2014.09.006
Bialk-Bielinska, A.; Caban, M.; Pieczynska, A.; Stepnowski, P.; Stolte, S. Mixture toxicity of six sulfonamides and their two transformation products to green algae Scenedesmus vacuolatus and duckweed Lemna minor. Chemosphere 2017, 173, 542−550. doi: 10.1016/j.chemosphere.2017.01.035
Adams, C.; Wang, Y.; Loftin, K.; Meyer, M. Removal of antibiotics from surface and distilled water in conventional water treatment processes. J. Environ. Eng. 2002, 128, 253−260. doi: 10.1061/(ASCE)0733-9372(2002)128:3(253)
Huber, M. M.; Korhonen, S.; Ternes, T. A.; Von, G. U. Oxidation of pharmaceuticals during water treatment with chlorine dioxide. Water Res. 2005, 39, 3607−3617. doi: 10.1016/j.watres.2005.05.040
Yu, F.; Li, Y.; Han, S.; Ma, J. Adsorptive removal of antibiotics from aqueous solution using carbon materials. Chemosphere 2016, 153, 365−385. doi: 10.1016/j.chemosphere.2016.03.083
Klavarioti, M.; Mantzavinos, D.; Kassinos, D. Removal of residual pharmaceuticals from aqueous systems by advanced oxidation processes. Environ. Int. 2009, 35, 402−417. doi: 10.1016/j.envint.2008.07.009
Mika, S.; Mohamed, C. N.; Anu, M. Advanced oxidation processes for the removal of natural organic matter from drinking water sources: a comprehensive review. J. Environ. Manage. 2018, 208, 56−76. doi: 10.1016/j.jenvman.2017.12.009
Abellán, M. N.; Bayarri, B.; Giménez, J.; Costa. J. Photocatalytic degradation of sulfamethoxazole in aqueous suspension of TiO2. Appl. Catal. B Environ. 2007, 74, 233−241. doi: 10.1016/j.apcatb.2007.02.017
Yang, H.; Li, G. Y.; An, T. C.; Gao, Y. P.; Fu, J. M. Photocatalytic degradation kinetics and mechanism of environmental pharmaceuticals in aqueous suspension of TiO2: a case of sulfa drugs. Catal. Today 2010, 153, 200−207. doi: 10.1016/j.cattod.2010.02.068
Długosz, M.; Żmudzki, P.; Kwiecień, A.; Szczubiałka, K.; Krzek, J.; Nowakowska, M. Photocatalytic degradation of sulfamethoxazole in aqueous solution using a floating TiO2-expanded perlite photocatalyst. J. Hazard. Mater. 2015, 298, 146−153. doi: 10.1016/j.jhazmat.2015.05.016
Zhu, W. Y.; Sun, F. Q.; Goei, R.; Zhou, Y. Facile fabrication of RGO-WO3 composites for effective visible light photocatalytic degradation of sulfamethoxazole. Appl. Catal. B Environ. 2017, 207, 93−102. doi: 10.1016/j.apcatb.2017.02.012
Mirzaei, A.; Yerushalmi, L.; Chen, Z.; Haghighat, F.; Guo, J. B. Enhanced photocatalytic degradation of sulfamethoxazole by zinc oxide photocatalyst in the presence of fluoride ions: optimization of parameters and toxicological evaluation. Water Res. 2018, 132, 241−251. doi: 10.1016/j.watres.2018.01.016
Trovó, A. G.; Nogueira, R. F. P.; Agüera, A.; Sirtori, C.; Fernandez-Alba, A. R. Photodegradation of sulfamethoxazole in various aqueous media: persistence, toxicity and photoproducts assessment. Chemosphere 2009, 77, 1292−1298. doi: 10.1016/j.chemosphere.2009.09.065
Du, J. S.; Guo, W. Q.; Wang, H. Z.; Yin, R. L.; Zheng, H. S.; Feng, X. C.; Che, D.; Ren, N. Q. Hydroxyl radical dominated degradation of aquatic sulfamethoxazole by Fe0/bisulfite/O2: kinetics, mechanisms, and pathways. Water Res. 2018, 138, 323−332. doi: 10.1016/j.watres.2017.12.046
Ge, L. K.; Zhang, P.; Halsall, C.; Li, Y. Y.; Chen, C. E.; Li, J.; Sun, H. L.; Yao, Z. W. The importance of reactive oxygen species on the aqueous phototransformation of sulfonamide antibiotics: kinetics, pathways, and comparisons with direct photolysis. Water Res. 2019, 149, 243−250. doi: 10.1016/j.watres.2018.11.009
Shah, S.; Hao, C. Quantum chemical investigation on photodegradation mechanisms of sulfamethoxypyridazine with dissolved inorganic matter and hydroxyl radical. J. Environ. Sci. 2017, 57, 85−92. doi: 10.1016/j.jes.2016.09.023
Yu, H.; Chen, J. W.; Xie, H. B.; Ge, P.; Kong, Q. W.; Luo, Y. Ferrate(VI) initiated oxidative degradation mechanisms clarified by DFT calculations: a case for sulfamethoxazole. Environ. Sci. Proc. Impacts 2017, 19, 370−378. doi: 10.1039/C6EM00521G
Zhang, H. M.; Wei, X. X.; Song, X. D.; Shah, S.; Chen, J. W.; Liu, J. H.; Hao, C.; Chen, Z. F. Photophysical and photochemical insights into the photodegradation of sulfapyridine in water: a joint experimental and theoretical study. Chemosphere 2018, 191, 1021−1027. doi: 10.1016/j.chemosphere.2017.10.036
Lee, J. E.; Choi, W.; Mhin, B. J.; Balasubramanian, K. Theoretical study on the reaction of OH radicals with polychlorinated dibenzo-p-dioxins. J. Phys. Chem. A 2004, 108, 607−614. doi: 10.1021/jp036084q
Altarawneh, M.; Kennedy, E. M.; Dlugogorski, B. Z.; Mackie, J. C. Computational study of the oxidation and decomposition of dibenzofuran under atmospheric conditions. J. Phys. Chem. A 2008, 112, 6960−6967. doi: 10.1021/jp800093j
Zeng, X. L.; Zhang, X. L.; Wang, Z. Y. Theoretical study on the OH-initiated oxidation mechanism of polyfluorinated dibenzo-p-dioxins under the atmospheric conditions. Chemosphere 2016, 144, 2036−2043. doi: 10.1016/j.chemosphere.2015.10.106
Zeng, X. L.; Chen, J.; Qu, R. J.; Pan, X. X.; Wang, Z. Y. The OH-initiated atmospheric chemical reactions of polyfluorinated dibenzofurans and polychlorinated dibenzofurans: a comparative theoretical study. Chemosphere 2017, 168, 10−17. doi: 10.1016/j.chemosphere.2016.10.062
Wang, Y.; Zeng, X. L. OH-initiated atmospheric oxidation mechanism of 1-chloropyrene: a theoretical study. Comput. Theoret. Chem. 2017, 1115, 144−150. doi: 10.1016/j.comptc.2017.06.011
Qu, R. J.; Liu, H. X.; Feng, M. B.; Yang, X.; Wang, Z. Y. Investigation on intramolecular hydrogen bond and some thermodynamic properties of polyhydroxylated anthraquinones. J. Chem. Eng. Data 2012, 57, 2442−2455. doi: 10.1021/je300407g
Shi, J. Q.; Qu, R. J.; Feng, M. B.; Wang, X. H.; Wang, L. S.; Yang, S. G.; Wang, Z. Y. Oxidative degradation of decabromodiphenyl ether (BDE 209) by potassium permanganate: reaction pathways, kinetics, and mechanisms assisted by density functional theory calculations. Environ. Sci. Technol. 2015, 49, 4209−4217. doi: 10.1021/es505111r
Qu, R. J.; Li, C. G.; Liu, J. Q.; Xiao, R. Y.; Pan, X. X.; Zeng, X. L.; Wang, Z. Y.; Wu, J. C. Hydroxyl radical based photocatalytic degradation of halogenated organic contaminants and paraffin on silica gel. Environ. Sci. Technol. 2018, 52, 7220−7229. doi: 10.1021/acs.est.8b00499
Zhao, J. M.; Yuan, Y.; Zhou, R. J. A molecular mechanism of Fenton oxidation degradation of m-xylene. Chin. J. Struct. Chem. 2018, 37, 1363−1370.
Cheng, X. L. Absorption and emission spectra of p-phenylbenzoyl methanthiol: methanol effect based on M06-2X calculations. Chin. J. Struct. Chem. 2019, 38, 476−482.
Zhang, L.; Chen, R.; Liang, G. M. Theoretical studies on the aminolysis of ester: effects of the catalysis and substituent to the reaction of methyl indole-3-acetate with ammonia. Chin. J. Struct. Chem. 2019, 38, 855−866.
Zhou, J.; Chen, J. W.; Liang, C. H.; Xie, Q.; Wang, Y. N.; Zhang, S. Y.; Qiao, X. L.; Li, X. H. Quantum chemical investigation on the mechanism and kinetics of PBDE photooxidation by OH: a case study for BDE-15. Environ. Sci. Technol. 2011, 45, 4839−4845. doi: 10.1021/es200087w
Zhao, Y.; Truhlar, D. G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215−241. doi: 10.1007/s00214-007-0310-x
Miertuš, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilization of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117−129. doi: 10.1016/0301-0104(81)85090-2
Miertuš, S.; Tomasi, J. Approximate evaluations of the electrostatic free energy and internal energy changes in solution processes. Chem. Phys. 1982, 65, 239−245. doi: 10.1016/0301-0104(82)85072-6
Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G. A.; Nakatsuji, H.; Caricato, M.; Li, X.; Hratchian, H. P.; Izmaylov, A. F.; Bloino, J.; Zheng, G.; Sonnenberg, J. L.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Vreven, T.; Montgomery, J. A.; Peralta, J. E.; Ogliaro, F.; Bearpark, M.; Heyd, J. J.; Brothers, E.; Kudin, K. N.; Staroverov, V. N.; Kobayashi, R.; Normand, J.; Raghavachari, K.; Rendell, A.; Burant, J. C.; Iyengar, S. S.; Tomasi, J.; Cossi, M.; Rega, N.; Millam, J. M.; Klene, M.; Knox, J. E.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Martin, R. L.; Morokuma, K.; Zakrzewski, V. G.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Dapprich, S.; Daniels, A. D.; Farkas, O.; Foresman, J. B.; Ortiz, J. V.; Cioslowski, J. Fox, D. J. Gaussian Inc., Wallingford, CT 2009, Gaussian 09, Revision A. 2.
Pilling, M. J.; Seakins, P. W. Reaction Kinetics. Oxford University Press Inc., New York 1999.
Haag, W. R.; Yao, C. C. D. Rate constants for reaction of hydroxyl radicals with several drinking water contaminants. Environ. Sci. Technol. 1992, 26, 1005−1013. doi: 10.1021/es00029a021
Monod, A.; Poulain, L.; Grubert, S.; Voisin, D.; Wortham, H. Kinetics of OH-initiated oxidation of oxygenated organic compounds in the aqueous phase: new rate constants, structure-activity relationships and atmospheric implications. Atmos. Environ. 2005, 39, 7667−7688. doi: 10.1016/j.atmosenv.2005.03.019
Olariu, R. I.; Klotz, B.; Barnes, I.; Becker, K. H.; Mocanu, R. FT-IR study of the ring-retaining products from the reaction of OH radicals with phenol, o-, m-, and p-cresol. Atmos. Environ. 2002, 36, 3685−3697. doi: 10.1016/S1352-2310(02)00202-9
Mezyk, S. P.; Neubauer, T. J.; Cooper, W. J.; Peller, J. R. Free-radical-induced oxidative and reductive degradation of sulfa drugs in water: absolute kinetics and efficiencies of hydroxyl radical and hydrated electron reactions. J. Phys. Chem. A 2007, 111, 9019−9024. doi: 10.1021/jp073990k

Figure 1 Molecular structures of sulfamethoxazole (SMX), sulfadiazine (SDZ) and sulfapyridine (SPY) and the numbering system of C atoms

Figure 2 OH addition reactions of SMX and their Gibbs free energies of activation (ΔrG≠, in kcal⋅mol−1) and Gibbs free energies of reaction (ΔrG, in kcal⋅mol−1) in aqueous solution at 298.15 K (The data in parentheses are the results for the reactions in gas phase)
Table 1. Calculated Frontier Molecular Orbital (FMO) Energies for Three Sulfonamides
| Sulfonamides | HOMO/a.u. | LUMO/a.u. |
| SMX+ | −0.32880 | −0.04519 |
| SMX | −0.27947 | −0.00930 |
| SMX− | −0.26225 | 0.00467 |
| SDZ+ | −0.32378 | −0.04379 |
| SDZ | −0.27771 | −0.02385 |
| SDZ− | −0.25920 | −0.00126 |
| SPY+ | −0.31495 | −0.04348 |
| SPY | −0.27878 | −0.01488 |
| SPY− | −0.25403 | 0.00465 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Overall Rate Constants (k) for OH Addition Reactions of Three Forms of SMX, SDZ and SPY at 298.15 K
| Reactions | k/M–1⋅s–1 |
| SMX+ + OH → SMX+-OH adducts | 4.75 × 106 |
| SMX + OH → SMX-OH adducts | 6.93 × 109 |
| SMX− + OH → SMX−-OH adducts | 1.42 × 1011 |
| SDZ+ + OH → SDZ+-OH adducts | 1.52 × 106 |
| SDZ + OH → SDZ-OH adducts | 2.00 × 1010 |
| SDZ− + OH → SDZ−-OH adducts | 3.93 × 1010 |
| SPY+ + OH → SPY+-OH adducts | 8.85 × 106 |
| SPY + OH → SPY-OH adducts | 1.99 × 109 |
| SPY− + OH → SPY−-OH adducts | 4.22 × 1011 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: