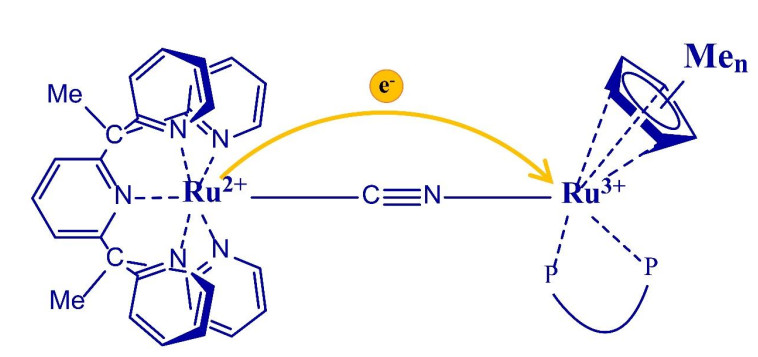
Figure 1.
Chemical structure of complexes 23+ (n = 0), 33+ (n = 1) and 43+ (n = 5)

The investigation on electron transfer process has attracted a lot of attention from chemists and physicists over the past decades[1-4], because understanding electron transfer process is very important in some critical issues such as designing artificial photosynthesis[5], exposing catalytic mechanisms[6], development of superconducting materials[4], design of molecular electronic devices[7, 8], etc. Mixed-valence (MV) complexes are ideal simple models for investigating electron transfer process[9-13]. Low-valent metals can be used as electron donors to transfer electrons to high-valent metal electron acceptor fragments. Using mixed valence model makes it easy to calculate the electron transfer rate and the activation energy of intervalence electron transfer[14, 15]. Among them, the investigation on dinuclear ruthenium is the most common[16-19], such as the Creutz-Taube ion[20]. Various bridges can be used to connect electron donors and acceptors, such as pyrazine[20], alkyne[21-31], cyanide bridges[16, 32-54], naphthalene[55-58], the organic bridge with redox activity[59, 60] and even mononuclear metals with multiple coordination sites[50, 54]. To date, it has been investigated how electron transfer process is influenced by the distance between the two metals[30], the energy barrier of electron transfer[54] and the cis-/trans-configuration. Cyanide is a short-range and asymmetric linear bridging ligand. The cyanide C-terminal metal feeds back much stronger electrons to the C≡N anti-orbital through the dπ orbital than the cyanide N-terminal metal does. For this reason, the cyanide C-terminal metal is often used as electron-donor and the cyanide N-terminal metal as electron-acceptor. The metal center can effectively transfer electrons through the cyanide bridge. Therefore, cyanide bridge is often used as a bridging ligand to investigate the electron transfer process in asymmetric mixed-valence dinuclear compounds.
Our group has carried out a systematic study on the regulation of electron transfer in cyanide bridged mixed-valence compounds by different methods, such as changing the electron donating ability of ligands, cyanide bridge orientation, cis-trans isomerism and so on[49, 50, 53, 54]. In previous studies, we found that changing the electron-donating ability of the ligand in the electron acceptor fragment has a more impact on the behavior of MMCT than in the electron-donor fragment[53]. To further investigate the influence of the electron-acceptor fragment's electron-accepting ability on the behavior of MMCT, we have synthesized and characterized three complexes [(PY5Me2)Ru(μ-CN)Ru(dppe)CpMen]2+ (22+, n = 0; 32+, n = 1; 42+, n = 5). As shown in Fig. 1, all the one-electron oxidation complexes N3+ (N = 2, 3, 4), which are obtained in situ by gradually adding cerium ammonium nitrate in acetonitrile solution of the N2+ complexes, exhibit a metal-to-metal charge transfer (MMCT) properties. The MMCT energy can be tuned by changing the number of methyl groups of cyclopentadiene located on the Ru center of the electron acceptor fragment, further supported by the time-dependent density functional theory (TD-DFT) calculations.
Vario MICRO elemental analyzer was used to detect the element content (C, H and N). Infrared (IR) spectra were collected using KBr pellets on a PerkinElmer Spectrum. UV-vis-NIR absorption spectroscopy was collected with the PerkinElmer Lambda 365 UV-vis-NIR spectrophotometer. The cyclic voltammetry curve was measured under argon by V3-Studio with methylene chloride as the solvent and 0.1 M (Bu4N) PF6 as the promoting electrolyte at a scan rate of 100 mVs-1. The electrolytic cell consists of glassy graphite as working electrode, platinum as counter electrode and Ag/AgCl as the reference electrode. Ferrocene was used to calibrate the potential. The single-crystal X-ray diffraction data for complexes 2 [PF6]2, 3 [PF6]2 and 4 [PF6]2 were collected on a Saturn 724+ CCD diffractometer equipped with graphite-monochromatic MoKa (λ = 0.71073 Å) radiation at 293 K. Complex 1 (PF6) was collected on a metal Jet D2+ diffractometer with graphite-monochromatic GaKα (λ = 1.3405 Å) radiation at 110 K. All structures were solved by intrinsic phasing methods using ShelXT-2018/3 and refined with ShelXL-2018/3[61], OLEX2[62] program package. The SQUEEZE program in the PLATON software was used[63].
All operations were performed under argon atmosphere using standard Schlenk technology, unless otherwise specified. PY5Me2[64], CpRu(dppe)Cl[65], CpMeRu(dppe)Cl[51] and CpMe5Ru(dppe)Cl[66] were prepared by the previous literature. Methanol, ethanol and dichloromethane are 50 ppm super-dry solvents purchased from Adamas. All other raw materials were purchased commercially and used without further purification.
At room temperature, RuCl3·3H2O (1.0 g, 3.82 mmol) was added to 300 mL of anhydrous ethanol. The mixture was refluxed at 88 ℃ for 6 hours, and then cooled to room temperature, to which PY5Me2 (1.7 g, 3.83 mmol) in 50 mL ethanol was added. The mixture was refluxed at 88 ℃ for 48 hours, cooled to room temperature and filtered to remove the precipitate. The filtrate was rotary evaporated to remove the solvent to obtain a pale yellow crude product, which was recrystallized with ethanol to obtain 1.4 g of pure product with a yield of 57.5%.
At room temperature, 10 equivalents of KCN (1.02 g, 15.7 mmol) were added to an aqueous solution of PY5Me2RuCl2 (1.0 g, 1.57 mmol). The mixture was refluxed at 110 ℃ for 2 hours, then cooled to room temperature, and an excess of NH4PF6 was added, resulting in a light green precipitate. The precipitate was filtered and dried in vacuum, giving 1.02 g light green product of 1[PF6] (yield 90.8%). The yellow crystals of 1[PF6] suitable for X-ray diffraction single-crystal structure analysis were obtained by slow diffusion of anhydrous ether into the DMF solution of 1[PF6] (75 mg, yield 81%).
Anal. Calcd. (%) for C30H25N6F6PRu·2H2O: C, 47.97; H, 3.86; N, 11.19. Found (%): C, 47.69/47.85; H, 4.02/4.04; N, 11.00/11.01. IR (νCN, KBr pellet): 2077 cm-1.
At room temperature, the compound PY5Me2RuCN [PF6] (50 mg, 0.07 mmol) was added to 1 equivalent of CpRu(dppe)Cl (42 mg, 0.07 mmol) in methanol (10 mL). The mixture was refluxed for 24 hours and cooled to room temperature. An excess of NH4PF6 was added and stirred for ten minutes to obtain a red precipitate. This precipitate was filtered and washed with a small amount of methanol and ether, giving the product of 2[PF6]2. The yellow crystals of 2[PF6]2 suitable for X-ray diffraction single-crystal structure analysis were obtained by slow diffusion of anhydrous ether into the dichloromethane solution of 2[PF6]2 (69 mg, yield 75%).
Anal. Calcd. (%) for C61H54F12N6P4Ru2·CH3CN: C, 51.30; H, 3.89; N, 6.69. Found (%): C, 50.78; H, 3.81; N, 6.90. IR (νCN, KBr pellet): 2093 cm-1.
At room temperature, the compound PY5Me2RuCN [PF6] (50 mg, 0.07 mmol) was added to 1 equivalent of Cp1Ru(dppe)Cl (43 mg, 0.07 mmol) in methanol (10 mL). The mixture was refluxed for 24 hours and cooled to room temperature. Then an excess of NH4PF6 was added and stirred for ten minutes to obtain a red precipitate which was filtered and washed with a small amount of methanol and ether, giving the product of 3[PF6]2. The yellow crystals of 3[PF6]2 suitable for X-ray diffraction single-crystal structure analysis were obtained by slow diffusion of anhydrous ether into the dichloromethane solution of 3[PF6]2 (75 mg, yield 81%).
Anal. Calcd. (%) for C62H56F12N6P4Ru2·CH3CN: C, 51.89; H, 3.99; N, 6.62. Found (%): C, 52.10/51.49; H, 3.98/4.01; N, 6.64/6.56. IR (νCN, KBr pellet): 2091 cm-1.
At room temperature, the compound PY5Me2RuCN [PF6] (50 mg, 0.07 mmol) was added to 1 equivalent of Cp5Ru (dppe) Cl (47 mg, 0.07 mmol) in methanol (10 mL), and the resulting mixture was refluxed for 24 hours and cooled to room temperature. An excess of NH4PF6 was added and stirred for ten minutes to obtain a red precipitate. The precipitate was filtered and washed with a small amount of methanol and ether, giving the product of 4[PF6]2. The yellow crystals of 4[PF6]2 suitable for X-ray diffraction single-crystal structure analysis were obtained by slow diffusion of anhydrous ether into the dichloromethane solution of 4[PF6]2 (55 mg, yield 53%).
Anal. Calcd. (%) for C66H64F12N6P4Ru2·H2O: C, 52.38; H, 4.37; N, 5.55. Found (%): C, 52.75/52.62; H, 4.78/4.89; N, 5.63/5.53. IR (νCN, KBr pellet): 2067 cm-1.
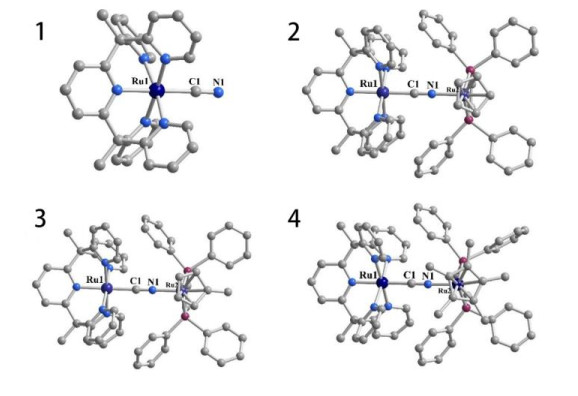
X-ray crystal structures of complexes 1[PF6] and 2[PF6]2~4[PF6]2 are shown in Fig. 2. The crystallographic data are summarized in Table 1. The space groups of compounds 1[PF6], 2[PF6]2, 3[PF6]2 and 4[PF6]2 are Pbcm, P
 DownLoad:
CSV
DownLoad:
CSV
| Complex | 1[PF6] | 2[PF6]2 [PF6]2·2CH2Cl2·CH3CN | 3[PF6]2 | 4[PF6]2·CH2Cl2 | |
| Empirical formula | C30H24.6F6N6PRu | C65H61Cl4F12N7P4Ru2 | C62H56F12N6P4Ru2 | C67H66Cl2F12N6P4Ru2 | |
| Formula weight | 715.20 | 1636.02 | 1439.07 | 1580.17 | |
| Temperature/K | 293(2) | 293(2) | 100.00(10) | 293(2) | |
| Crystal system | Orthorhombic | Triclinic | Monoclinic | Monoclinic | |
| Space group | Pbcm | P |
P21/m | P21/n | |
| a/Å | 9.02510(10) | 11.1761(14) | 11.4028(4) | 14.3242(4) | |
| b/Å | 17.5909(3) | 14.0233(7) | 23.5113(6) | 24.0896(6) | |
| c/Å | 21.2449(4) | 23.9935(7) | 13.4498(6) | 21.0103(5) | |
| α/° | 90 | 86.469(3) | 90 | 90 | |
| β/° | 90 | 89.574(3) | 113.502(5) | 102.308(2) | |
| γ/° | 90 | 66.624(4) | 90 | 90 | |
| Volume/Å3 | 3372.83(9) | 3444.6(3) | 3306.7(2) | 7083.3(3) | |
| Z | 4 | 2 | 2 | 4 | |
| ρcalcg/cm3 | 1.408 | 1.577 | 1.445 | 1.482 |
|
| μ/mm-1 | 0.574 | 0.765 | 3.480 | 0.668 | |
| F(000) | 1438 | 1648 | 1452 | 3200 | |
| Crystal size/mm3 | 0.32×0.11×0.08 | 0.31×0.28×0.20 | 0.31×0.15×0.11 | 0.11×0.11×0.08 | |
| Radiation | MoKα | MoKα | Micro-focus metaljet | MoKα | |
| 2θ range/° | 4.632 to 52.732 | 3.5 to 52.742 | 6.23 to 121.62 | 3.366 to 52.744 | |
| Reflections collected | 46444 | 48982 | 47679 | 106915 | |
| Independent reflections | 3442 | 14083 | 7671 | 14482 | |
| Data/restraints/parameters | 3442/6/223 | 14083/0/850 | 7671/64/455 | 14482/0/845 | |
| Goodness-of-fit on F2 | 1.099 | 1.042 | 1.080 | 1.035 | |
| Final R indexes (I > 2σ(I)) | R = 0.0311, wR = 0.0778 | R = 0.0920, wR = 0.2372 | R = 0.0376, wR = 0.1006 | R = 0.0550, wR = 0.1510 | |
| Final R indexes (all data) | R = 0.0332, wR = 0.0790 | R = 0.1237, wR = 0.2748 | R = 0.0442, wR = 0.1056 | R = 0.0697, wR = 0.1617 |
DownLoad:
CSV
| 1[PF6] | 2[PF6]2 | 3[PF6]2 | 4[PF6]2 | |
| Ru(1)–C(1) | 2.013(3) | 1.990(6) | 1.993(4) | 2.026(4) |
| C(1)–N(1) | 1.134(4) | 1.144(8) | 1.148(5) | 1.162(5) |
| N(1)–Ru(2) | 2.039(5) | 2.055(3) | 2.089(3) | |
| Ru(1)–N(av.) | 2.077(18) | 2.076(5) | 2.083(4) | 2.087(4) |
| Ru(2)–P(av.) | 2.2661(18) | 2.2697(7) | 2.3095(11) | |
| Ru(2)–Cp | 1.864 | 1.857 | 1.891 | |
| Ru(1)···Ru(2) | 5.155 | 5.191 | 5.277 | |
| Ru(1)–C(1)–N(1) | 178.3(3) | 174.0(6) | 175.4(3) | 178.1(4) |
| Ru(2)–N(1)–C(1) | 175.0(5) | 178.7(3) | 177.5 (3) |
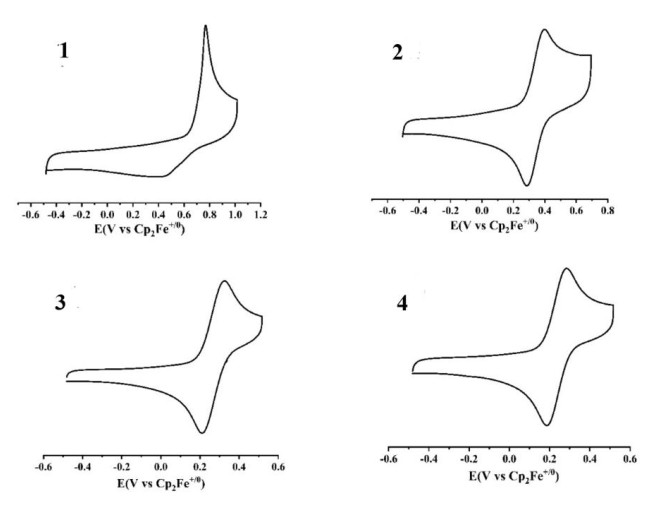
The cyclic voltammetry of the four compounds in dichloromethane all showed one reversible redox wave. The results are shown in Fig. 3 and Table 2. The cyclic voltammetry of complexes 2[PF6]2, 3[PF6]2 and 4[PF6]2 each shows one reversible redox wave at 0.342, 0.267 and 0.234 V, respectively, which could be attributed to CpMen(dppe)RuII/CpMen(dppe)RuIII and is higher than the similar monomer[67] based on the previous paper[53]. The redox wave of the mononuclear complex 1[PF6]2 exhibits one redox wave assigned to (PY5Me2)RuII/(PY5Me2)RuIII. However, the same redox wave for (PY5Me2)RuII/(PY5Me2)RuIII in the three dinuclear complexes was not observed, which may be due to its too higher potential position. From Table 3, it can be found that with the increase of the number of methyl groups on cyclopentadiene the redox wave position moves to a lower potential from 2[PF6]2 and 3[PF6]2 to 4[PF6]2.
 DownLoad:
CSV
DownLoad:
CSV
| Complex | L | E1/2/V |
| 1[PF6] | 0.59 | |
| 2[PF6]2 | Cp | 0.342 |
| 3[PF6]2 | CpMe | 0.267 |
| 4[PF6]2 | CpMe5 | 0.234 |
Infrared spectroscopy is an excellent way to characterize cyanide bridged compounds. The contraction vibration of cyanide is easy to observe in infrared spectroscopy, and the cyanide stretching ν(CN) position can help us judge the connection rigidity and electrons feedback situation of the compounds. Compared with the position of the terminal cyanide signal of the mononuclear compound [PY5Me2RuCN]+ (2077 cm-1), the position of the bridged cyanide band of the dinuclear compounds 22+ (2093 cm-1) and 32+ (2091 cm-1) has a blue shift due to the rigid restriction of the cyanide N-coordinated Ru on the movement of the bridging cyanide. But for compound 42+, the pentamethylcyclopentadiene ligand has a stronger electron-donating ability. It promotes the transfer of d orbital electron of the nitrogen-terminal Ru fragment to the π anti-bond orbital of the cyanide bridge to form a feedback π bond, which reduces the cyanide energy level and results in the absorption peak redshift. With the enhancement of the electron-donating ability of the ligand on the C-terminal metal, the redshift of the cyanide vibration frequency is often observed, but it is rare to reduce the cyanide vibration energy through the feedback electron from the N-terminal metal.
DownLoad:
CSV
| Complex | L | ν(cm-1) |
| 1[PF6] | 2077 | |
| 2[PF6]2 | Cp | 2093 |
| 3[PF6]2 | CpMe | 2091 |
| 4[PF6]2 | CpMe5 | 2067 |
UV-VIS-NIR absorption spectroscopy is the most effective method for studying electron transfer. According to the strongest absorption position, absorption intensity and half-width of the MMCT absorption peak, the strength of the electron transfer of the compound can be investigated. In order to study the influence of the electron-donating ability of the acceptor terminal ligand on the MMCT, we used the acetonitrile solution of cerium ammonium nitrate to gradually oxidize the three dinuclear compounds to obtain mixedvalence compounds in situ. Their absorption spectra were measured, as shown in Figs. 4 and 5. The absorption peak from 27500 cm-1 to 22000 cm-1 for each of the mixed-valence compounds 23+, 33+ and 43+ obtained in situ is attributed to the metal-to-ligand electron transfer (MLCT) from Ru2+ to the PY5Me2 ligand[68], and the new absorption peak from 16000 cm-1 to 9000 cm-1 is attributed to the metal-to-metal electron transfer (MMCT)[53]. For MLCT absorption peaks, the maxima absorption peaks of the three compounds 22+, 32+ and 42+ before oxidation are basically the same, all at about 25000 cm-1. After one-electron oxidation, it can be observed that the positions of the MLCT maxima absorption peaks of the three dinuclear mixed valence Ru compounds 23+, 33+ and 43+each exhibits a significant blue shift, and the absorption intensity is also significantly weakened. This is because that from 22+, 32+ and 42+ to 23+, 33+ and 43+ the electron-donating ability of RuII in the (PY5Me2)RuII fragment weakens due to the electron withdraw effect of the CpMen(dppe)RuIII.
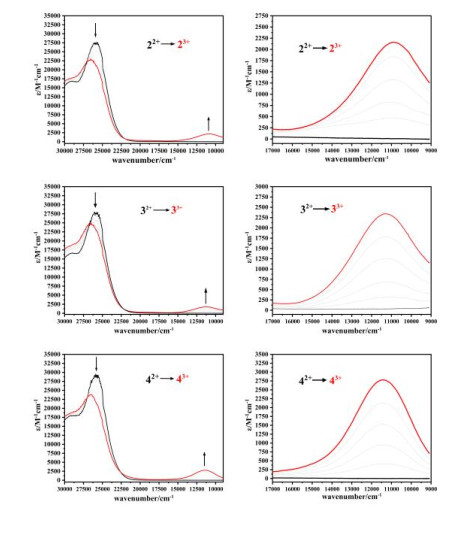
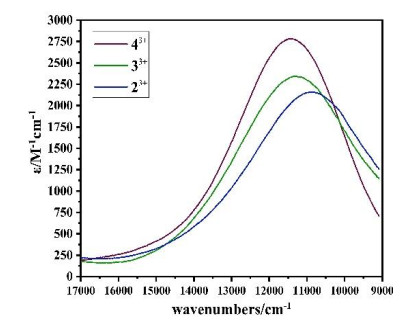
For the MMCT absorption peak, with the increase of the electron-donating ability of the substituted group from Cp, CpMe to CpMe5, the MMCT absorption peaks of the three mixed-valence compounds each shows a blue shift (10953 cm-1 for 23+, 11274 cm-1 for 33+ and 11442 cm-1 for 43+), and the absorption intensity increases (2125M-1 cm-1→2324 M-1 cm-1→2786 M-1 cm-1). This is because that the electron-accepting ability of RuIII in the CpMen(dppe)RuIII fragment decreases as the electron-donating ability increases from Cp, CpMe to CpMe5, resulting in the increase of the RuII→RuIII MMCT energy from 23+, 33+ to 43+, strongly supported by the TD-DFT calculation.
The electronic coupling constant Hab of complexes 23+~43+ was calculated using the Hush-Mullikan equation (Eq. 1)[9], with the results listed in Table 5. In the equation, ν1/2 is the bandwidth at half-intensity of the MMCT band maximum vmax, and εmax and rAB represent the molar extinction coefficient and the through space intermetallic distance, respectively. As shown in Table 5, the electronic coupling constant Hab gradually increases from 23+, 33+ to 43+. This may be understood by the fact that as the number of methyl groups of Cp increases the mixed valence state [RuII-CN-RuIII] becomes more and more stable, resulting in the more and more MMCT energy from [RuII-CN-RuIII] to [RuIII-CN-RuII] from 23+, 33+ to 43+, and the Hab increases correspondingly. According to the calculation results, the one-electron oxidation products 23+, 33+ and 43+ belong to the class II mixed valence compounds.
|
|
(Eq. 1) |
DownLoad:
CSV
| Complex | λmax/cm-1 | εmax/M-1·cm-1 | Hab |
| 2[PF6]3 | 10953 | 2125 | 1235 |
| 3[PF6]3 | 11274 | 2324 | 1293 |
| 4[PF6]3 | 11442 | 2786 | 1479 |
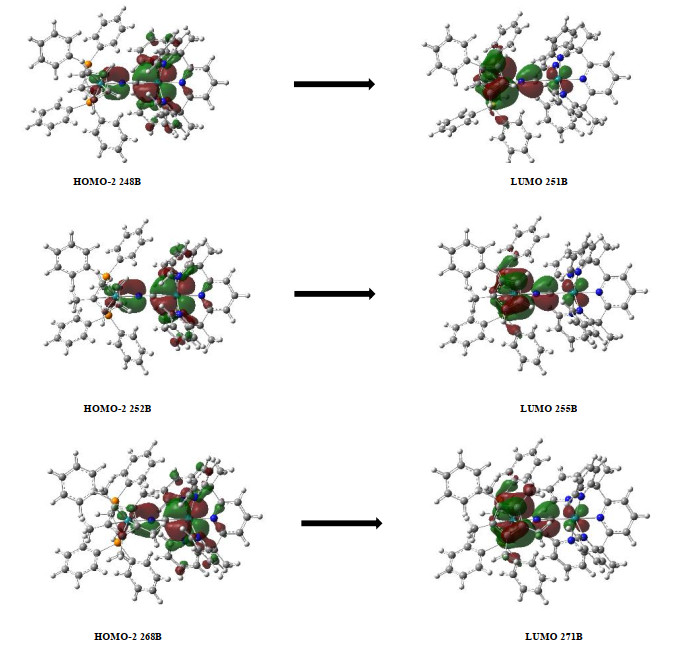
To continue studying the influence of electron acceptor orbital changes on the properties of MMCT, we performed TD-DFT calculations using B3LYP/lanl2dz[69, 70] for the three MV compounds. As shown in Table 6, the spin electron density of the three MV compounds is mainly localized on RuIII, further indicating that the single-electron oxidation products of the three compounds belong to the class Ⅱ mixed-valence compounds. Due to the decrease of electron-accepting ability of the acceptor fragments from 23+, 33+ to 43+, the RuII → RuIII MMCT gets to be more difficult, resulting in an increase of the spin electron density on the acceptor metal center. For MMCT, the calculation results are basically consistent with the experimental data, as shown in Table 7. The metal donor HOMO orbitals and acceptor LUMO orbitals of the three MV compounds are shown in Fig. 6. The major contribution for the MMCT absorption band of complex 23+ comes from molecular orbital 248B to 251B. For compound 33+, the MMCT absorption peak is mainly derived from molecular orbital 252B to 255B. For compound 43+, the MMCT absorption peak mainly comes from the molecular orbitals from 268B to 271B. The LUMO orbitals of the three compounds are almost localized on Ru3+, while the HOMO ones are localized on Ru2+, which indicates that the three compounds all belong to the class II mixed-valence compounds, consistent with the measured UV-Vis-NIR absorption spectral results.
DownLoad:
CSV
| Ru1 | Ru2 | |
| 23+ | 0.072983 | 0.761033 |
| 33+ | 0.072559 | 0.779743 |
| 43+ | 0.042988 | 0.832436 |
DownLoad:
CSV
| Complex | Measured (cm-1) | Calculated (cm-1) |
| 23+ | 10953 | 12469 |
| 33+ | 11274 | 12407 |
| 43+ | 11442 | 13755 |
In summary, we have synthesized and characterized a mononuclear Ru fragment and three cyanido-bridged dinuclear Ru compounds 22+, 32+ and 42+. All one-electron oxidation complexes 23+, 33+ and 43+ obtained in situ show a MMCT absorption band in the NIR range. The MMCT energy increases as the number of methyl groups on the cyclopentadiene of the cyanido-nitrogen coordinated Ru metal increases, supported by the DFT/TDDFT calculations. Furthermore, the UV-vis-NIR absorption spectra and TDDFT calculations show that all the single-electron oxidation compounds belong to class Ⅱ mixed-valence compounds. This work shows that slight modifications to the ligand on the N-terminal metal center can tune the MMCT properties of the mixed valence compounds.
Marcus, R. A. Chemical and electrochemical electron-transfer theory. Annu. Rev. Phys. Chem. 1964, 15, 155–196. doi: 10.1146/annurev.pc.15.100164.001103
Hush, N. S. Homogeneous and heterogeneous optical and thermal electron transfer. Electrochim. Acta 1968, 13, 1005–1023. doi: 10.1016/0013-4686(68)80032-5
Rafiq, S.; Scholes, G. D. From fundamental theories to quantum coherences in electron transfer. J. Am. Chem. Soc. 2019, 141, 708–722. doi: 10.1021/jacs.8b09059
Aubrey, M. L.; Wiers, B. M.; Andrews, S. C.; Sakurai, T.; Reyes-Lillo, S. E.; Hamed, S. M.; Yu, C. J.; Darago, L. E.; Mason, J. A.; Baeg, J. O.; Grandjean, F.; Long, G. J.; Seki, S.; Neaton, J. B.; Yang, P.; Long, J. R. Electron delocalization and charge mobility as a function of reduction in a metal-organic framework. Nat. Mater. 2018, 17, 625–632. doi: 10.1038/s41563-018-0098-1
Levanon, H.; Norris, J. R. The photoexcited triplet state and photosynthesis. Chem. Rev. 1978, 78, 185–198. doi: 10.1021/cr60313a001
Kochi, J. K. Electron-transfer mechanisms for organometallic intermediates in catalytic reactions. Acc. Chem. Res. 2002, 7, 351–360.
Nitzan, A.; Ratner, M. A. Electron transport in molecular wire junctions. Science 2003, 300, 1384–9. doi: 10.1126/science.1081572
Wenger, O. S. Photoswitchable mixed valence. Chem. Soc. Rev. 2012, 41, 3772–9. doi: 10.1039/c2cs15339d
Heckmann, A.; Lambert, C. Organic mixed-valence compounds: a playground for electrons and holes. Angew Chem. Int. Ed. Engl. 2012, 51, 326–92. doi: 10.1002/anie.201100944
Kang, M. T.; Meng, M.; Tan, Y. N.; Cheng, T.; Liu, C. Y. Tuning the electronic coupling and electron transfer in Mo2 donor-acceptor systems by variation of the bridge conformation. Chemistry 2016, 22, 3115–26. doi: 10.1002/chem.201504033
Zhu, G. Y.; Meng, M.; Tan, Y. N.; Xiao, X.; Liu, C. Y. Electronic coupling between two covalently bonded dimolybdenum units bridged by a naphthalene group. Inorg. Chem. 2016, 55, 6315–22. doi: 10.1021/acs.inorgchem.6b01021
Cheng, T.; Xiao, X.; Zhang, L.; Liu, C. Y.; Wang, L. L.; Meng, M.; Zhao, F.; Wang, H.; Ji, L. N. Photoinduced delta electron transfer in phenylene bridged Mo2 dimers. Phys. Chem. Chem. Phys. 2017, 19, 1740–1745. doi: 10.1039/C6CP07582G
Zhu, G. Y.; Qin, Y.; Meng, M.; Mallick, S.; Gao, H.; Chen, X.; Cheng, T.; Tan, Y. N.; Xiao, X.; Han, M. J.; Sun, M. F.; Liu, C. Y. Crossover between the adiabatic and nonadiabatic electron transfer limits in the landau-zener model. Nat. Commun. 2021, 12, 456. doi: 10.1038/s41467-020-20557-7
Lancaster, K.; Odom, S. A.; Jones, S. C.; Thayumanavan, S.; Marder, S. R.; Bredas, J. L.; Coropceanu, V.; Barlow, S. Intramolecular electron-transfer rates in mixed-valence triarylamines: measurement by variable-temperature ESR spectroscopy and comparison with optical data. J. Am. Chem. Soc. 2009, 131, 171–23.
Slenkamp, K. M.; Lynch, M. S.; Van Kuiken, B. E.; Brookes, J. F.; Bannan, C. C.; Daifuku, S. L.; Khalil, M. Investigating vibrational anharmonic couplings in cyanide-bridged transition metal mixed valence complexes using two-dimensional infrared spectroscopy. J. Chem. Phys. 2014, 140, 084505. doi: 10.1063/1.4866294
Oviedo, P. S.; Pieslinger, G. E.; Cadranel, A.; Baraldo, L. M. Exploring the localized to delocalized transition in non-symmetric bimetallic ruthenium polypyridines. Dalton Trans. 2017, 46, 15757–15768. doi: 10.1039/C7DT02422C
Tang, J. H.; Shao, J. Y.; He, Y. Q.; Wu, S. H.; Yao, J.; Zhong, Y. W. Transition from a metal-localized mixed-valence compound to a fully delocalized and bridge-biased electrophore in a ruthenium-amine-ruthenium tricenter system. Chemistry 2016, 22, 10341–5. doi: 10.1002/chem.201601806
Zhong, Y. W.; Gong, Z. L.; Shao, J. Y.; Yao, J. Electronic coupling in cyclometalated ruthenium complexes. Coord. Chem. Rev. 2016, 312, 22–40. doi: 10.1016/j.ccr.2016.01.002
Shao, J. Y.; Gong, Z. L.; Zhong, Y. W. Bridged cyclometalated diruthenium complexes for fundamental electron transfer studies and multi-stage redox switching. Dalton Trans. 2018, 47, 23–29. doi: 10.1039/C7DT04168C
Creutz, C.; Taube, H. Direct approach to measuring the franck-condon barrier to electron transfer between metal ions. J. Am. Chem. Soc. 1969, 91, 3988–3989. doi: 10.1021/ja01042a072
Field, L. D.; Turnbull, A. J.; Turner, P. Acetylide-bridged organometallic oligomers via the photochemical metathesis of methyl-iron(II) complexes. J. Am. Chem. Soc. 2002, 124, 3692–702. doi: 10.1021/ja011105k
Venkatesan, K.; Blacque, O.; Berke, H. Organometallic manganese complexes as scaffolds for potential molecular wires. Dalton Trans. 2007, 1091–100.
Olivier, C.; Kim, B.; Touchard, D.; Rigaut, S. Redox-active molecular wires incorporating ruthenium(II) σ-arylacetylide complexes for molecular electronics. Organometallics 2008, 27, 509–518. doi: 10.1021/om700779x
Benameur, A.; Brignou, P.; Di Piazza, E.; Hervault, Y. M.; Norel, L.; Rigaut, S. Redox-active ruthenium(II) σ-arylacetylide wires for molecular electronics incorporating insulating chains. New J. Chem. 2011, 35.
Luo, L.; Benameur, A.; Brignou, P.; Choi, S. H.; Rigaut, S.; Frisbie, C. D. Length and temperature dependent conduction of ruthenium-containing redox-active molecular wires. J. Phys. Chem. C 2011, 115, 19955–19961. doi: 10.1021/jp207336v
Egler-Lucas, C.; Blacque, O.; Venkatesan, K.; Lopez-Hernandez, A.; Berke, H. Dinuclear and mononuclear chromium acetylide complexes. Eur. J. Inorg. Chem. 2012, 2012, 1536–1545. doi: 10.1002/ejic.201100929
Lissel, F.; Fox, T.; Blacque, O.; Polit, W.; Winter, R. F.; Venkatesan, K.; Berke, H. Stepwise construction of an iron-substituted rigid-rod molecular wire: targeting a tetraferra-tetracosa-decayne. J. Am. Chem. Soc. 2013, 135, 4051–60. doi: 10.1021/ja400078c
Lissel, F.; Blacque, O.; Venkatesan, K.; Berke, H. Structural and electronic variations of sp/sp2 carbon-based bridges in di- and trinuclear redox-active iron complexes bearing Fe(diphosphine)2x (x = i, ncs) moieties. Organometallics 2015, 34, 408–418. doi: 10.1021/om500602m
Lissel, F.; Schwarz, F.; Blacque, O.; Riel, H.; Lörtscher, E.; Venkatesan, K.; Berke, H. Organometallic single-molecule electronics: tuning electron transport through x(diphosphine)2FeC4Fe(diphosphine)2x building blocks by varying the Fe-X-Au anchoring scheme from coordinative to covalent. J. Am. Chem. Soc. 2014, 136, 14560–14569. doi: 10.1021/ja507672g
Zheng, Q.; Hampel, F.; Gladysz, J. A. Longitudinally extended molecular wires based upon PtC⋮CC⋮CC⋮CC⋮C repeat units: iterative syntheses of functionalized linear PtC8Pt, PtC8PtC8Pt, and PtC8PtC8PtC8Pt assemblies. Organometallics 2004, 23, 589–5899. doi: 10.1021/om0342659
Semenov, S. N.; Blacque, O.; Fox, T.; Venkatesan, K.; Berke, H. Electronic communication in dinuclear C(4)-bridged tungsten complexes. J. Am. Chem. Soc. 2010, 132, 3115–27. doi: 10.1021/ja909764x
Nihei, M.; Ui, M.; Yokota, M.; Han, L.; Maeda, A.; Kishida, H.; Okamoto, H.; Oshio, H. Two-step spin conversion in a cyanide-bridged ferrous square. Angew Chem. Int. Ed. Engl. 2005, 44, 6484–7. doi: 10.1002/anie.200502216
Nihei, M.; Sekine, Y.; Suganami, N.; Nakazawa, K.; Nakao, A.; Nakao, H.; Murakami, Y.; Oshio, H. Controlled intramolecular electron transfers in cyanide-bridged molecular squares by chemical modifications and external stimuli. J. Am. Chem. Soc. 2011, 133, 3592–600. doi: 10.1021/ja109721w
Hoshino, N.; Iijima, F.; Newton, G. N.; Yoshida, N.; Shiga, T.; Nojiri, H.; Nakao, A.; Kumai, R.; Murakami, Y.; Oshio, H. Three-way switching in a cyanide-bridged [CoFe] chain. Nat. Chem. 2012, 4, 921–6. doi: 10.1038/nchem.1455
Jiao, C. Q.; Meng, Y. S.; Yu, Y.; Jiang, W. J.; Wen, W.; Oshio, H.; Luo, Y.; Duan, C. Y.; Liu, T. Effect of intermolecular interactions on metal-to-metal charge transfer: a combined experimental and theoretical investigation. Angew Chem. Int. Ed. Engl. 2019, 58, 17009–17015. doi: 10.1002/anie.201909495
Albores, P.; Slep, L. D.; Weyhermuller, T.; Baraldo, L. M. Fine tuning of the electronic coupling between metal centers in cyano-bridged mixed-valent trinuclear complexes. Inorg. Chem. 2004, 43, 6762–73. doi: 10.1021/ic0493649
Albores, P.; Slep, L. D.; Eberlin, L. S.; Corilo, Y. E.; Eberlin, M. N.; Benitez, G.; Vela, M. E.; Salvarezza, R. C.; Baraldo, L. M. From monomers to geometry-constrained molecules: one step further toward cyanide bridged wires. Inorg. Chem. 2009, 48, 11226–35. doi: 10.1021/ic901710x
Cadranel, A.; Albores, P.; Yamazaki, S.; Kleiman, V. D.; Baraldo, L. M. Efficient energy transfer via the cyanide bridge in dinuclear complexes containing Ru(II) polypyridine moieties. Dalton Trans. 2012, 41, 5343–50. doi: 10.1039/c2dt11869f
Pieslinger, G. E.; Albores, P.; Slep, L. D.; Coe, B. J.; Timpson, C. J.; Baraldo, L. M. Communication between remote moieties in linear Ru-Ru-Ru trimetallic cyanide-bridged complexes. Inorg. Chem. 2013, 52, 2906–17. doi: 10.1021/ic302173g
Pieslinger, G. E.; Albores, P.; Slep, L. D.; Baraldo, L. M. Class III delocalization in a cyanide-bridged trimetallic mixed-valence complex. Angew Chem. Int. Ed. Engl. 2014, 53, 1293–6. doi: 10.1002/anie.201307025
Pieslinger, G. E.; Aramburu-Troselj, B. M.; Cadranel, A.; Baraldo, L. M. Influence of the electronic configuration in the properties of d6-d5 mixed-valence complexes. Inorg. Chem. 2014, 53, 8221–9. doi: 10.1021/ic5002539
Cadranel, A.; Oviedo, P. S.; Pieslinger, G. E.; Yamazaki, S.; Kleiman, V. D.; Baraldo, L. M.; Guldi, D. M. Trapping intermediate mlct states in low-symmetry {Ru(bpy)} complexes. Chem. Sci. 2017, 8, 7434–7442. doi: 10.1039/C7SC02670F
Cadranel, A.; Oviedo, P. S.; Albores, P.; Baraldo, L. M.; Guldi, D. M.; Hodak, J. H. Electronic energy transduction from {Ru(py)4} chromophores to Cr(III) luminophores. Inorg. Chem. 2018, 57, 3042–3053. doi: 10.1021/acs.inorgchem.7b02799
Aramburu-Troselj, B. M.; Oviedo, P. S.; Pieslinger, G. E.; Hodak, J. H.; Baraldo, L. M.; Guldi, D. M.; Cadranel, A. A hole delocalization strategy: photoinduced mixed-valence mlct states featuring extended lifetimes. Inorg. Chem. 2019, 58, 10898–10904. doi: 10.1021/acs.inorgchem.9b01254
Aramburu-Troselj, B. M.; Oviedo, P. S.; Ramirez-Wierzbicki, I.; Baraldo, L. M.; Cadranel, A. Inversion of donor-acceptor roles in photoinduced intervalence charge transfers. Chem. Commun. (Camb) 2019, 55, 7659–7662. doi: 10.1039/C9CC03483H
Oviedo, P. S.; Pieslinger, G. E.; Baraldo, L. M.; Cadranel, A.; Guldi, D. M. Coexistence of mlct excited states of different symmetry upon photoexcitation of a single molecular species. J. Phys. Chem. C 2019, 123, 3285–3291.
Dominguez, S. E.; Pieslinger, G. E.; Sanchez-Merlinsky, L.; Baraldo, L. M. Does geometry matter? Effect of the ligand position in bimetallic ruthenium polypyridine siblings. Dalton Trans. 2020, 49, 4125–4135. doi: 10.1039/D0DT00040J
Sheng, T.; Vahrenkamp, H. Long range metal-metal interactions along Fe−NC−Ru−CN−Fe chains. Eur. J. Inorg. Chem. 2004, 2004, 1198–1203.
Ma, X.; Lin, C. S.; Zhu, X. Q.; Hu, S. M.; Sheng, T. L.; Wu, X. T. An unusually delocalized mixed-valence state of a cyanidometal-bridged compound induced by thermal electron transfer. Angew Chem. Int. Ed. Engl. 2017, 56, 1605–1609. doi: 10.1002/anie.201610855
Yang, Y. Y.; Zhu, X. Q.; Hu, S. M.; Su, S. D.; Zhang, L. T.; Wen, Y. H.; Wu, X. T.; Sheng, T. L. Different degrees of electron delocalization in mixed valence Ru-Ru-Ru compounds by cyanido-/isocyanido-bridge isomerism. Angew Chem. Int. Ed. Engl. 2018, 57, 14046–14050. doi: 10.1002/anie.201806157
Li, S. H.; Liu, Y.; Yang, Y. Y.; Zhang, Y. X.; Xu, Q. D.; Hu, S. M.; Wu, X. T.; Sheng, T. L. Syntheses, crystal structures and mmct properties of cyanide-bridged binuclear Ru-Fe complexes. Polyhedron 2019, 173.
Su, S. D.; Zhu, X. Q.; Wen, Y. H.; Zhang, L. T.; Yang, Y. Y.; Lin, C. S.; Wu, X. T.; Sheng, T. L. A diruthenium-based mixed spin complex Ru2 (5+) (s = 1/2)-CN-Ru2 (5+) (s = 3/2). Angew Chem. Int. Ed. Engl. 2019, 58, 15344–15348. doi: 10.1002/anie.201909097
Zhang, L. T.; Zhu, X. Q.; Hu, S. M.; Zhang, Y. X.; Su, S. D.; Yang, Y. Y.; Wu, X. T.; Sheng, T. L. Influence of ligand substitution at the donor and acceptor center on mmct in a cyanide-bridged mixed-valence system. Dalton Trans. 2019, 48, 7809–7816. doi: 10.1039/C9DT01303B
Yang, Y. Y.; Zhu, X. Q.; Launay, J. P.; Hong, C. B.; Su, S. D.; Wen, Y. H.; Wu, X. T.; Sheng, T. L. Electron transfer process in mixed valence compounds with low-lying energy bridge in different oxidation states. Angew Chem. Int. Ed. Engl. 2020, 60, 4804–4814.
Hatanaka, T.; Ohki, Y.; Kamachi, T.; Nakayama, T.; Yoshizawa, K.; Katada, M.; Tatsumi, K. Naphthalene and anthracene complexes sandwiched by two {(Cp*)Fe(I)} fragments: strong electronic coupling between the Fe(I) centers. Chem. Asian J. 2012, 7, 1231–42. doi: 10.1002/asia.201101037
Schnöckelborg, E. M.; Hartl, F.; Langer, T.; Pöttgen, R.; Wolf, R. Redox-active, dinuclear sandwich compounds [Cp*Fe(μ-l)FeCp*] (l = naphthalene and anthracene). European Journal of Inorg. Chem. 2012, 2012, 1632–1638. doi: 10.1002/ejic.201200001
Malberg, J.; Lupton, E.; Schnöckelborg, E. M.; de Bruin, B.; Sutter, J.; Meyer, K.; Hartl, F.; Wolf, R. Synthesis and electronic structure of dissymmetrical, naphthalene-bridged sandwich complexes [Cp′Fe(μ-c10h8)mcp*]x (x = 0, +1; m = Fe, Ru; Cp′ = η5-c5h2-1, 2, 4-tbu3; Cp* = η5-C5Me5). Organometallics 2013, 32, 6040–6052. doi: 10.1021/om4005862
Herrmann, D.; Rodl, C.; de Bruin, B.; Hartl, F.; Wolf, R. Synthesis, electronic structure and redox properties of the diruthenium sandwich complexes [Cp*Ru(mu-C10H8)RuCp*](x) (x = 0, 1+; Cp* = C5Me5; C10H8 = naphthalene). Dalton Trans. 2018, 47, 11058–11069. doi: 10.1039/C8DT02003E
Ibanez, S.; Poyatos, M.; Peris, E. Mono and dimetallic pyrene-imidazolylidene complexes of iridium(III) for the deuteration of organic substrates and the C-C coupling of alcohols. Dalton Trans. 2016, 45, 14154–9. doi: 10.1039/C6DT02942F
Carter, A.; Mason, A.; Baker, M. A.; Bettler, D. G.; Changas, A.; McMillen, C. D.; Tapu, D. Janus-type bis(malonhc) and its zwitterionic gold and silver metal complexes. Organometallics 2017, 36, 1867–1872. doi: 10.1021/acs.organomet.7b00206
Sheldrick, G. M. Crystal structure refinement with shelxl. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. doi: 10.1107/S2053229614024218
Dolomanov, O. V.; Bourhis, L. J.; Gildea, R. J.; Howard, J. A. K.; Puschmann, H. Olex2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. doi: 10.1107/S0021889808042726
Spek, A. L. Structure validation in chemical crystallography. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 148–55. doi: 10.1107/S090744490804362X
Bechlars, B.; D'Alessandro, D. M.; Jenkins, D. M.; Iavarone, A. T.; Glover, S. D.; Kubiak, C. P.; Long, J. R. High-spin ground states via electron delocalization in mixed-valence imidazolate-bridged divanadium complexes. Nat. Chem. 2010, 2, 362–8. doi: 10.1038/nchem.585
Gluyas, J. B. G.; Brown, N. J.; Farmer, J. D.; Low, P. J. Optimised syntheses of the half-sandwich complexes FeCl(dppe)Cp*, FeCl(dppe)Cp, RuCl(dppe)Cp*, and RuCl(dppe)Cp. Aust. J. Chem. 2017, 70, 113–119. doi: 10.1071/CH16322
Bruce, M. I.; Ellis, B. G.; Low, P. J.; Skelton, B. W.; White, A. H. Syntheses, structures, and spectro-electrochemistry of {Cp*(PP)Ru}C⋮CC⋮C{Ru(PP)Cp*} (pp = dppm, dppe) and their mono- and dications. Organometallics 2003, 22, 3184–3198. doi: 10.1021/om030015g
Perkins, G. J.; Bruce, M. I.; Skelton, B. W.; White, A. H. A new precursor for organo-osmium complexes. Inorg. Chim. Acta 2006, 359, 2644–2649. doi: 10.1016/j.ica.2005.09.070
Ohzu, S.; Ishizuka, T.; Kotani, H.; Kojima, T. Reactivity of a Ru(III)-hydroxo complex in substrate oxidation in water. Chem. Commun. (Camb) 2014, 50, 15018–21. doi: 10.1039/C4CC07488B
Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. doi: 10.1063/1.464913
Hay, P. J.; Wadt, W. R. Ab initio effective core potentials for molecular calculations. Potentials for k to Au including the outermost core orbitals. J. Chem. Phys. 1985, 82, 299–310. doi: 10.1063/1.448975

Figure 2 X-ray crystal structures of complexes 1~4 ([PF6]- ions, hydrogen atoms, acetonitrile and dichloromethane molecules have been removed for easy observation). Ru, dark blue; N, blue; P, wine red; C, grey

Figure 3 Cyclic voltammograms of 12+~42+ in a 0.10M dichloromethane solution of Bu4NPF6 at a scan rate of 100 mV·s-1 vs (Cp2Fe)+/0

Figure 4 UV-Vis-NIR absorption spectra of complexes 2~4 oxidized by the addition of ammonium ceric nitrate in acetonitrile

Figure 5 MMCT absorption spectra of complexes 2~4 oxidized by adding ammonium ceric nitrate in acetonitrile

Figure 6 Molecular orbital diagrams of HOMO-2 (248B) and LUMO (251B) for 2 (above), HOMO-2 (252B) and LUMO (255B) for 3 (middle), HOMO-2 (268B) and LUMO (271B) for 4 (bottom). The isosurface value is 0.02 au
Table 1. Test Condition, Structure Refine and Crystallographic Data for Compounds 1[PF6], 2[PF6]2, 3[PF6]2 and 4[PF6]2
| Complex | 1[PF6] | 2[PF6]2 [PF6]2·2CH2Cl2·CH3CN | 3[PF6]2 | 4[PF6]2·CH2Cl2 | |
| Empirical formula | C30H24.6F6N6PRu | C65H61Cl4F12N7P4Ru2 | C62H56F12N6P4Ru2 | C67H66Cl2F12N6P4Ru2 | |
| Formula weight | 715.20 | 1636.02 | 1439.07 | 1580.17 | |
| Temperature/K | 293(2) | 293(2) | 100.00(10) | 293(2) | |
| Crystal system | Orthorhombic | Triclinic | Monoclinic | Monoclinic | |
| Space group | Pbcm | P |
P21/m | P21/n | |
| a/Å | 9.02510(10) | 11.1761(14) | 11.4028(4) | 14.3242(4) | |
| b/Å | 17.5909(3) | 14.0233(7) | 23.5113(6) | 24.0896(6) | |
| c/Å | 21.2449(4) | 23.9935(7) | 13.4498(6) | 21.0103(5) | |
| α/° | 90 | 86.469(3) | 90 | 90 | |
| β/° | 90 | 89.574(3) | 113.502(5) | 102.308(2) | |
| γ/° | 90 | 66.624(4) | 90 | 90 | |
| Volume/Å3 | 3372.83(9) | 3444.6(3) | 3306.7(2) | 7083.3(3) | |
| Z | 4 | 2 | 2 | 4 | |
| ρcalcg/cm3 | 1.408 | 1.577 | 1.445 | 1.482 |
|
| μ/mm-1 | 0.574 | 0.765 | 3.480 | 0.668 | |
| F(000) | 1438 | 1648 | 1452 | 3200 | |
| Crystal size/mm3 | 0.32×0.11×0.08 | 0.31×0.28×0.20 | 0.31×0.15×0.11 | 0.11×0.11×0.08 | |
| Radiation | MoKα | MoKα | Micro-focus metaljet | MoKα | |
| 2θ range/° | 4.632 to 52.732 | 3.5 to 52.742 | 6.23 to 121.62 | 3.366 to 52.744 | |
| Reflections collected | 46444 | 48982 | 47679 | 106915 | |
| Independent reflections | 3442 | 14083 | 7671 | 14482 | |
| Data/restraints/parameters | 3442/6/223 | 14083/0/850 | 7671/64/455 | 14482/0/845 | |
| Goodness-of-fit on F2 | 1.099 | 1.042 | 1.080 | 1.035 | |
| Final R indexes (I > 2σ(I)) | R = 0.0311, wR = 0.0778 | R = 0.0920, wR = 0.2372 | R = 0.0376, wR = 0.1006 | R = 0.0550, wR = 0.1510 | |
| Final R indexes (all data) | R = 0.0332, wR = 0.0790 | R = 0.1237, wR = 0.2748 | R = 0.0442, wR = 0.1056 | R = 0.0697, wR = 0.1617 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Selected Bond Lengths (Å) and Bond Angles (°) for Compounds 1~4
| 1[PF6] | 2[PF6]2 | 3[PF6]2 | 4[PF6]2 | |
| Ru(1)–C(1) | 2.013(3) | 1.990(6) | 1.993(4) | 2.026(4) |
| C(1)–N(1) | 1.134(4) | 1.144(8) | 1.148(5) | 1.162(5) |
| N(1)–Ru(2) | 2.039(5) | 2.055(3) | 2.089(3) | |
| Ru(1)–N(av.) | 2.077(18) | 2.076(5) | 2.083(4) | 2.087(4) |
| Ru(2)–P(av.) | 2.2661(18) | 2.2697(7) | 2.3095(11) | |
| Ru(2)–Cp | 1.864 | 1.857 | 1.891 | |
| Ru(1)···Ru(2) | 5.155 | 5.191 | 5.277 | |
| Ru(1)–C(1)–N(1) | 178.3(3) | 174.0(6) | 175.4(3) | 178.1(4) |
| Ru(2)–N(1)–C(1) | 175.0(5) | 178.7(3) | 177.5 (3) |
下载: 导出CSV
Table 3. Electrochemical Data (vs Cp2Fe+/0) for Complexes 1~4 in 0.10 M DCM Solution of Bu4NPF6 at a Scan Rate of 100 mV·s-1
| Complex | L | E1/2/V |
| 1[PF6] | 0.59 | |
| 2[PF6]2 | Cp | 0.342 |
| 3[PF6]2 | CpMe | 0.267 |
| 4[PF6]2 | CpMe5 | 0.234 |
下载: 导出CSV
Table 4. CN Stretching Frequencies for Complexes 1(PF6)~4(PF6)2
| Complex | L | ν(cm-1) |
| 1[PF6] | 2077 | |
| 2[PF6]2 | Cp | 2093 |
| 3[PF6]2 | CpMe | 2091 |
| 4[PF6]2 | CpMe5 | 2067 |
下载: 导出CSV
Table 5. MMCT Transition Energies and Electronic Coupling Constant for All the Mixed-valence Complexes
| Complex | λmax/cm-1 | εmax/M-1·cm-1 | Hab |
| 2[PF6]3 | 10953 | 2125 | 1235 |
| 3[PF6]3 | 11274 | 2324 | 1293 |
| 4[PF6]3 | 11442 | 2786 | 1479 |
下载: 导出CSV
Table 6. Mulliken Spin Density of Mixed-valence Species
| Ru1 | Ru2 | |
| 23+ | 0.072983 | 0.761033 |
| 33+ | 0.072559 | 0.779743 |
| 43+ | 0.042988 | 0.832436 |
下载: 导出CSV
Table 7. Comparison of the Measured and the Calculated MMCT Energies of 23+, 33+ and 43+
| Complex | Measured (cm-1) | Calculated (cm-1) |
| 23+ | 10953 | 12469 |
| 33+ | 11274 | 12407 |
| 43+ | 11442 | 13755 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: