Citation:
Zhen LIANG, Wen-Li YAN, Hong-Mei LI, Ying LI, Rong ZHANG. Studies on the Molecular Mechanism between HDAC8 and Inhibitory in Different Bioactivities by Molecular Docking and MD Simulations[J]. Chinese Journal of Structural Chemistry,
2021, 40(10): 1298-1308.
doi:
10.14102/j.cnki.0254–5861.2011–3126

Studies on the Molecular Mechanism between HDAC8 and Inhibitory in Different Bioactivities by Molecular Docking and MD Simulations
English
Studies on the Molecular Mechanism between HDAC8 and Inhibitory in Different Bioactivities by Molecular Docking and MD Simulations
Abstract:
HDAC8 is an important target for the treatment of many cancers and other diseases. To develop potent and selective HDAC8 inhibitors, molecular docking and molecular dynamics (MD) simulations were employed for investigation of the mechanism of HDAC8 inhibitions containing hydroxamic acid group. Compound 1 with high activity and compound 2 with low activity were selected for comparative study. Compound 1 formed a stronger chelation with Zn ion and was more stable in the HDAC8 pocket than compound 2. Residues HIS-180, ASP-178, ASP-267, and GLY-140 played a critical role in securing the position of compound 1. Both the head and tail of compound 1 formed strong hydrogen bonds with ASP-178, facilitating the ZBG of compound 1 close to the Zn ion so that they formed permanent chelation during the simulation period. The Cap group of the compounds with branch and long chains was advantageous to form interaction with active pocket opening. What's more, based on the results of this study, three innovative recommendations for the design of highly active HDAC8 inhibitors were presented, which will be useful for the development of new HDAC8 inhibitors.
-
1. INTRODUCTION
Histone deacetylases (HDACs) are 'eraser' enzymes that cleave acetate from acetylated e-amino groups of lysine in histones and other proteins[1-3]. In humans, there are 18 kinds of HDACs divided into class I, II, III and IV[4]. The HDAC8 belongs to class I and is found in both nucleus and cytoplasm. It possesses 377 amino acids (aa, 42 kDa)[5-8] and is a zincdependent HDAC[9, 10].
HDAC8 is mainly studied as a cancer target and is highly expressed in a variety of cancer tissues compared to normal tissues[11-14]. Inhibiting HDAC8 is effective in the treatment of cancers containing T-cell lymphoma[15], Myeloid leukemia[16], Colon cancer[17], Breast cancer[18], Gastric carcinoma[19], Neuroblastoma[20], Hepatocellular carcinoma[21], Urothelial cancer[22], etc. Besides, inhibition of HDAC8 can fight the parasitic infections[23, 24], viral infections[25, 26] and Cornelia DE Lange syndrome (CdLS)[27].
Till date, only six HDAC inhibitors have successfully passed clinical trials. However, the inhibitors interact with all classes of HDACs and lead to adverse effects. Therefore, it is important to develop specific HDAC8 inhibitors. Most compounds containing hydroxamic acid group have a good inhibitory effect on HDAC8. Ingham et al.[28] have found a highly potent HDAC8 inhibitor namely OJI-1 (HDAC8 IC50 = 0.8 nM), a kind of hydroxamic acid. It was identified as the most selective and potent HDAC8 inhibitor till now over all other HDACs (HDAC1 to HDAC7 and HDAC9).
Currently, no specific HDAC8 inhibitor has been marketed. HDAC8 inhibitors known have either low activity or high toxicity. Therefore, it is still of great significance to continue to develop HDAC8 specific inhibitors with high activity and low toxic and side effects on human body. In addition, the specific inhibitory mechanism of inhibitors containing hydroxamic acid group on HDAC8 is still not clear, which needs further study.
Nowadays, computational aid drug design (CADD) is a successful technique in drug discovery[29, 30]. Recently, Zhou et al. have used molecular docking and MD simulation to study the mechanism by which three series of novel urushiol derivatives interact with HDAC8[31]. Uba et al. have reached a conclusion that 1T64 (HDAC8 in complex with TSA) and similar crystal structures may be good candidates for HDAC8 structural dynamics studies and inhibitor design[32].
In this work, a high active compound and a low active compound were selected for molecular docking and MD simulations. The results of the two compounds were compared to reveal the mystery of inhibition of HDAC8 by the compounds containing hydroxamic acid group.
2. MATERIALS AND METHOD
2.1 Materials
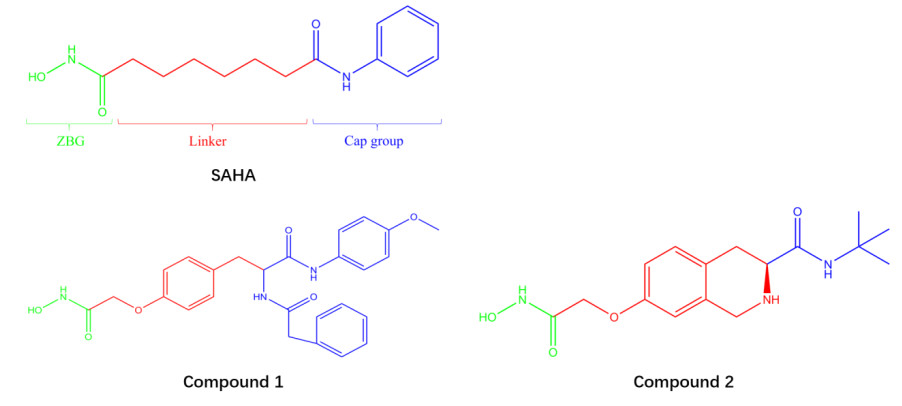
In this work, the highest active compound 1 and the lowest active compound 2 were selected among 61 HDAC8 inhibitors containing hydroxamic acid group and synthesized by Zhang et al.[33-35] (The structures are shown in Fig. 1). Compound 1 (IC50: 0.063 uM) is 23.5 times more active than the SAHA (Vorinostat, IC50: 1.48 uM[36]) and 193 times more active than compound 2. The receptor is the crystal structure of HDAC8 (1T64) downloaded from Protein Data Bank (https://www.rcsb.org/).
Figure 1

2.2 Molecular docking
AutoDock Vina 1.1.2 package software was performed for molecular docking. The receptor is the crystal structure of HDAC8, and the ligand is compounds 1 and 2 in the database. The PDBQT format for the receptor and ligand molecules was prepared in MGL tools 1.5.6. The maps of binding site were calculated with 15 × 15 × 15 grid points of 1 Å spacing.35. The molecular docking was repeated 5 times to get 100 conformations of the ligands. The ligands with optimal conformation in them were selected as the initial conformation of the molecular dynamics simulation[37].
2.3 Molecular dynamic simulation
The molecular dynamics simulation was carried out using Gromacs5.1.2 software. The GROMACS 96 force field is used to calculate proteins and compounds. The system was placed into a periodic cube with conditions of 298 K and 101.3 kPa and solvated with simple point charge (SPC) water molecules. Sodium ions were added to the box to satisfy the electrical neutrality condition. The calculation is regulated and conserved by isothermal-isochoric ensemble (NVT) and isothermal-isobaric ensemble (NPT). Once the equilibrium calculation of 100 ps was completed under the NVT and NPT ensembles, a 50 ns molecular MD simulation was performed[38].
3. RESULTS AND DISCUSSION
3.1 Docking analysis
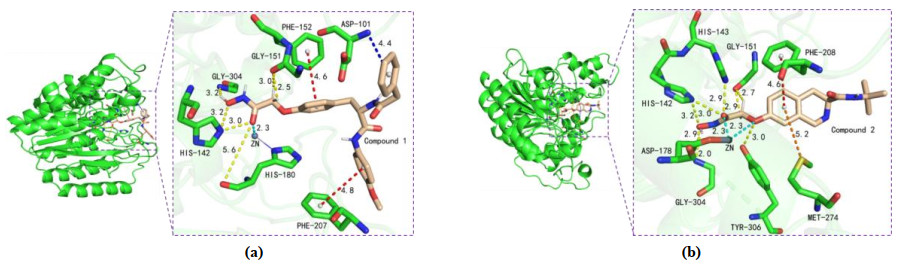
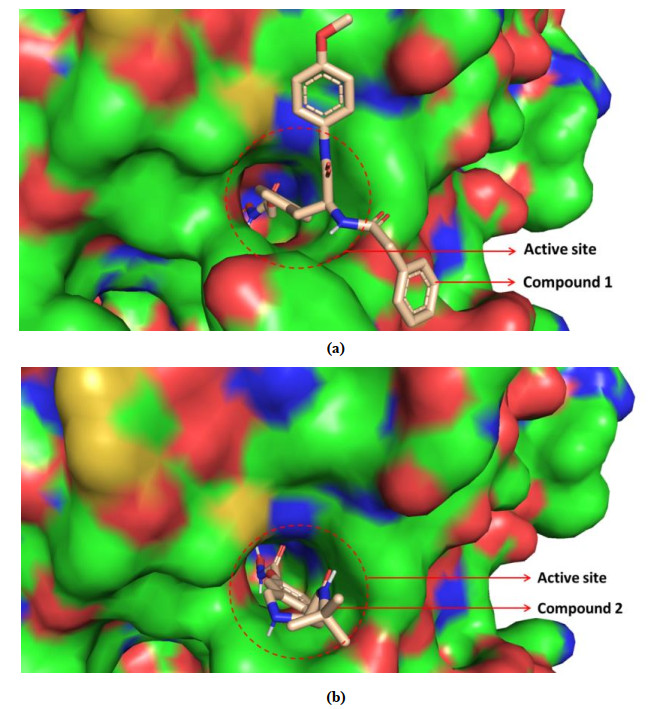
HDAC8 active site consists of a narrow and long hydrophobic tunnel, and a Zn ion is in the end of the hydrophobic tunnel[39]. The docking results of compounds 1 and 2 with HDAC8 are presented in Fig. 2, providing the mechanism about the compounds inhibiting HDAC8. The optimal docking score of compounds 1 and 2 was –8.8 and –7.8 kJ/mol, respectively. This indicated that the structure of compound 1 was more favorable for binding to the active site than that of compound 2. The compounds both chelated with Zn ion in active pocket, which was crucial mechanism of compounds containing hydroxamic acid group[9]. The ZBG (Zn ion Binding Group), Linker and Cap groups of compound 1 (Fig. 1) interacted with the residues of the active site. The Cap group of compound 1 was branched and interacted with both sides of the pocket opening, so compound 1 was embedded in HDAC8 like an iron nail, which fixed its position and made it be longer chelation with Zn ion. This is clearly and intuitively shown in Fig. 3a. However, only compound 2's ZBG and Linker interacted with the receptor. The Cap group had no interaction with the pocket opening due to its lack of branching and short length. Thereby, it is like an embroidery needle and easy to get out of the pocket (Fig. 3b).
Figure 2
 Figure 2. (a) Interactions between the active site of HDAC8 (color: green) and compound 1 (color: wheat). Metal chelating bond, H-bonds, π-π T-shaped and π-anion are depicted as cyan, yellow, red and blue dotted lines, respectively. (b) The Zn ion is shown as dark grey sphere. Interactions between the active site of HDAC8 (color: green) and compound 2 (color: wheat). Metal chelating bonds, H-bonds, π-π T-shaped and π-sulfur are depicted as cyan, yellow, red and orange dotted lines, respectively. The Zn ion is shown as dark grey sphere
Figure 2. (a) Interactions between the active site of HDAC8 (color: green) and compound 1 (color: wheat). Metal chelating bond, H-bonds, π-π T-shaped and π-anion are depicted as cyan, yellow, red and blue dotted lines, respectively. (b) The Zn ion is shown as dark grey sphere. Interactions between the active site of HDAC8 (color: green) and compound 2 (color: wheat). Metal chelating bonds, H-bonds, π-π T-shaped and π-sulfur are depicted as cyan, yellow, red and orange dotted lines, respectively. The Zn ion is shown as dark grey sphereFigure 3
 Figure 3. (a) Conformation of compound 1 bound to the active site. (b) Conformation of compound 2 bound to the active site
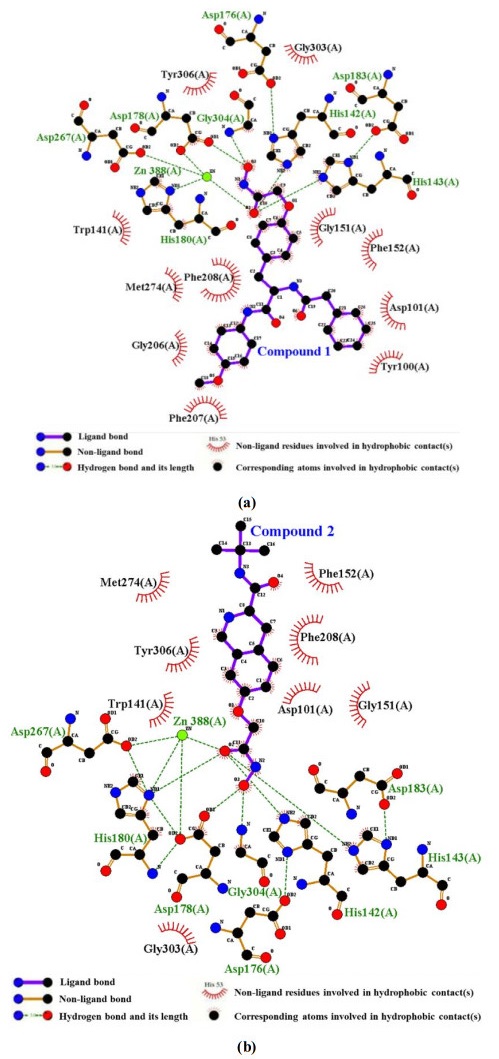
Figure 3. (a) Conformation of compound 1 bound to the active site. (b) Conformation of compound 2 bound to the active siteThe hydrophobic interaction of the compounds with active site is shown in Fig. 4. The compounds had hydrophobic interactions with many residues. The ZBG of the compounds formed a lot of hydrogen bonds (H-bonds), while the Linker and the Cap group formed lots of hydrophobic interactions. Moreover, significantly more residues formed hydrophobic bonds with compound 1 than 2, suggesting that hydrophobic interaction was also the reason why compound 1 binds more strongly to HDAC8 than 2.
Figure 4
 Figure 4. Hydrophobic interaction of complex system (a) HDAC8-compound 1 and (b) HDAC8-compound 2
Figure 4. Hydrophobic interaction of complex system (a) HDAC8-compound 1 and (b) HDAC8-compound 23.2 Analysis of MD simulations
3.2.1 Binding mode of complex system
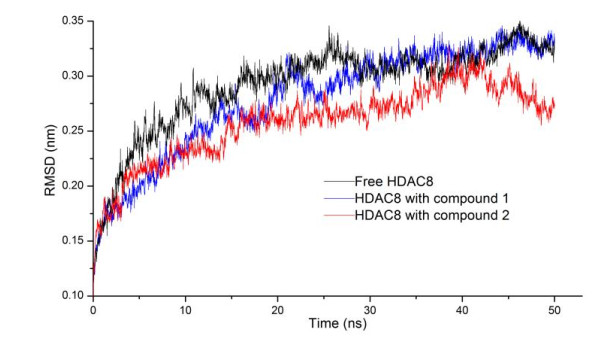
The root mean square deviation (RMSD) was applied for investigating the stability of complexes and conformational change. As shown in Fig. 5, RMSD value of HDAC8 with compounds 1 and 2 were lower than the RMSD of HDAC8 without ligand, which showed that the two compounds stabilized HDAC8 and formed more stable complexes with HDAC8. RMSD of HDAC8 with compound 1 had little fluctuation and HDAC8 with compound 2 exhibits larger fluctuation after the preliminary stability (25~50 ns), which manifested that the interaction between compound 1 and HDAC8 was stronger than that between compound 2 and HDAC8.
Figure 5
 Figure 5. RMSDs of HDAC8
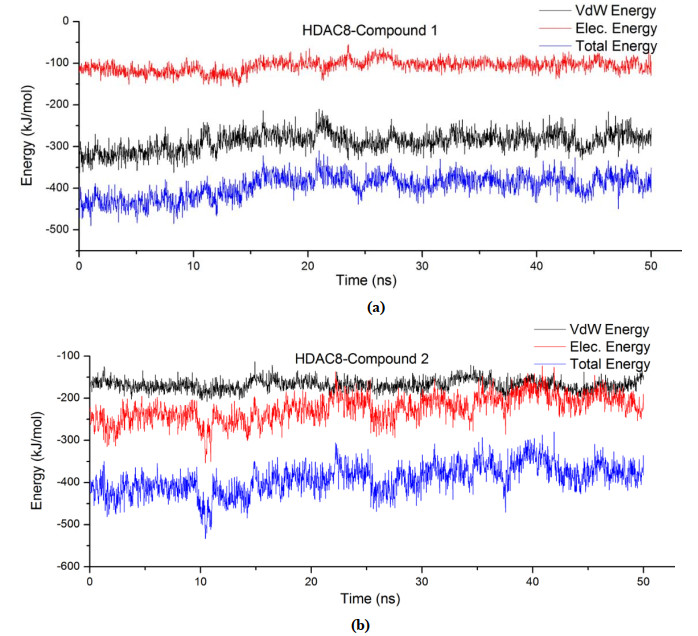
Figure 5. RMSDs of HDAC8The binding free energy of the two compounds to the receptor is shown in Fig. 6. The Van der Waals energy (VdW energy) accounted for a greater proportion of the binding free energy between compound 1 and HDAC8. However, the electrostatic energy (Elec energy) made up a higher proportion of the binding free energy between compound 2 and HDAC8. The Vdw energy of compound 1 was three times that of the Elec energy, and the Elec energy of compound 2 was slightly greater than the VdW energy. In addition, the binding energy of compound 1 to the receptor was stable within 50 ns with little fluctuation, whereas it is opposite for 2. What's more, the binding energy of compound 1 was generally lower than that of 2. Therefore, 1 has stronger interactions with HDAC8. The conclusion is consistent with that of RMSD.
Figure 6
 Figure 6. Fluctuation of binding free energy of complex (a) HDAC8-compound 1 and (b) HDAC8-compound 2
Figure 6. Fluctuation of binding free energy of complex (a) HDAC8-compound 1 and (b) HDAC8-compound 2Binding energy contribution values indicated the importance of residues interacting with the ligand. Generally, the smaller it is, the greater the contribution to the combination is. It is bad for the combination if it is positive. Binding energy contribution values of the Zn ion and 9 residues are in Table 1. Binding energy contributions of Zn ion was –63.6 kJ/mol in the interaction between compound 1 and HDAC8, and it is 48.9 kJ/mol in the interaction between compound 2 and HDAC8, which revealed that the Zn ion had strong interactions with compound 1. On the contrary, it is repulsive to compound 2, which is bad for it to combine with compound 2. Chelation of the inhibitor with Zn ion was vital to inhibit the activity of HDAC8, so compound 1 was much more active than 2.
Table 1
 Table 1. Binding Energy Contributions between HDAC8 Residues and Compounds 1 and 2
Table 1. Binding Energy Contributions between HDAC8 Residues and Compounds 1 and 2 DownLoad:
CSV
DownLoad:
CSV
Residues and ions of HDAC8 Binding energy of HDAC8 and Compound 1 (kJ/mol) Residues and ions of HDAC8 Binding energy of HDAC8 and Compound 2 (kJ/mol) ZN-388 –197.506 ZN-388 48.927 PHE-208 –63.600 ASP-101 –52.187 HIS-180 –16.439 ASP-272 –34.813 MET-274 –14.043 ASP-267 –29.639 PHE-207 –11.505 ASP-178 –23.193 PHE-152 –10.415 ASP-29 –21.349 TRP-141 –9.116 GLU-148 –20.505 TYR-306 –6.959 ASP-183 –20.450 ASP-101 –6.828 ASP-233 –19.657 LYS-202 –5.891 ASP-87 –18.916 SER-150 –5.427 ASP-157 –18.713 3.2.2 Interaction between ligands and HDAC8
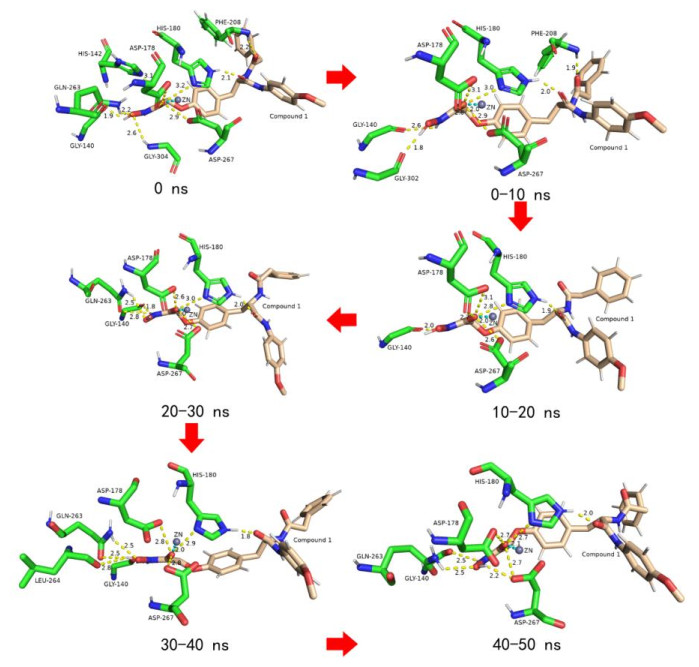
HIS-180, ASP-178 and ASP-267 chelated with Zn ion in HDAC8 to fix the position of Zn ion[40]. Interestingly, the O atom of the hydroxamic acid group of compound 1 not only chelated with Zn ion but also formed strong H-bonds with the three residues (Fig. 7). On the other side relative to the O atom, a H atom formed a constant H-bond with the O atom on GLY-140. At the cap group of compound 1, the O atom of the imide group had strong H-bonds with the H atom of imidazole ring of HIS-180. All the H-bonds did not break during the entire simulation period, indicating that the force was firm. It was the indestructible H-bonds that bonded to HDAC8 in all directions like iron chains, binding the ligand and receptor tightly together. In addition, compound 1 also formed a temporary H-bond with PHE-208, GLN-263, GLY-303, etc., further fixing the position of compound 1 in the active pocket. Therefore, compound 1 strongly inhibits the catalytic activity of HDAC8.
Figure 7
 Figure 7. Variation of HDAC8-compound 1 in the active sites during the MD trajectory
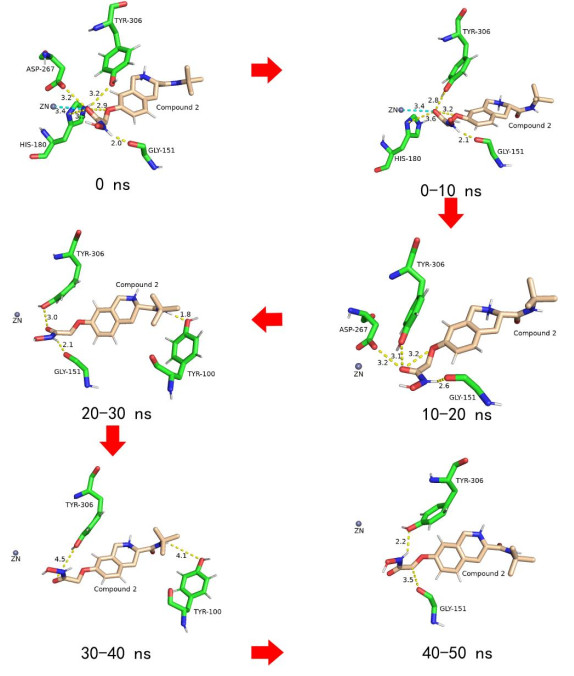
Figure 7. Variation of HDAC8-compound 1 in the active sites during the MD trajectoryCompound 2 interacted with HDAC8 rather than compound 1 with HDAC8 (Fig. 8). During the simulation period, TYR-306, His180 and GLY-151 residues formed H-bonds with compound 2 for a long time and played a role in fixing the position of the hydroxamic acid group of compound 2. However, GLY-151 and TYR-306 far from the Zn ion resulted in the O atom on the carbonyl group of hydroxamic acid group also far away from it, so it was difficult to bond with the Zn ion. And the Cap group of compound 2 did not interact with receptor and was easy to move, which further damaged the stability of the ZBG. Therefore, compound 2 was so unstable that it was easy to leave the active pocket, resulting in its low activity.
Figure 8
 Figure 8. Variation of HDAC8-compound 2 in the active sites during the MD trajectory
Figure 8. Variation of HDAC8-compound 2 in the active sites during the MD trajectory3.2.3 Interaction with Zn ion
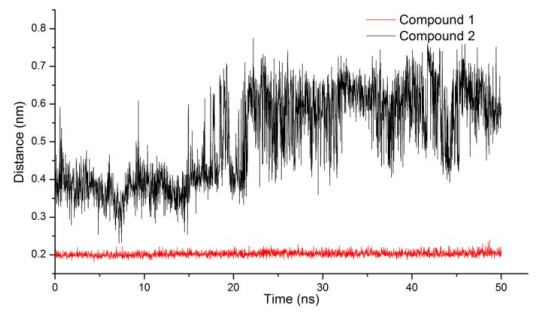
The literatures showed that the main mechanism of HDAC8 inhibitor containing hydroxamic acid group is the chelation with Zn ion[15, 41, 42]. During the simulation period, only can the O atom on the carbonyl group of hydroxamic acid group formed chelation with the Zn ion. Therefore, we figured out that the distance between the two atoms varies within 50 ns (Fig. 9). Firstly, the distance between the O atom of compound 1 and Zn ion maintained a steady level and small values (around 0.2 nm) in the 50 ns. This indicated that the interaction between compound 1 and the Zn ion was too firm to damage, while the distance between the O atom of compound 2 and Zn ion was greater than 0.4 nm. Such distance was difficult to chelate with Zn ion. What's more, this distance was larger in the later simulation period.
Figure 9
 Figure 9. Distance between Zn and the O atom on the carbonyl group of hydroxamic acid group
Figure 9. Distance between Zn and the O atom on the carbonyl group of hydroxamic acid group3.2.4 Analysis of H-bonds
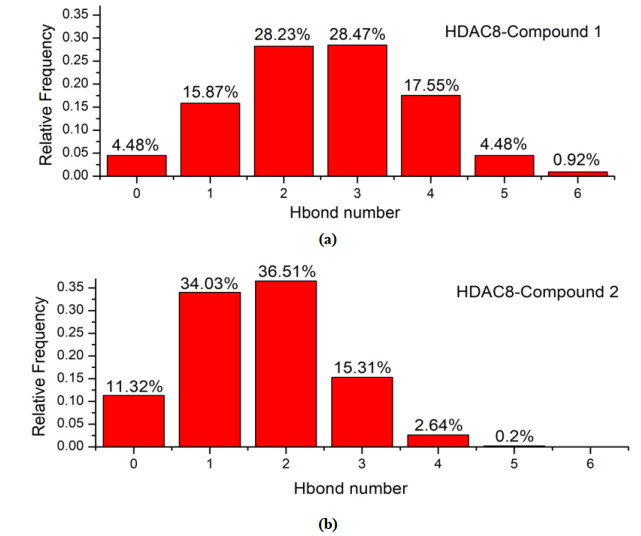
H-bonds played an important role in the interaction between the ligands and receptor. In general, the more H-bonds, the more stable the complex. The probability of the compounds forming H-bonds with the HDAC8 is shown in Fig. 10. There were at most six H-bonds formed between compound 1 and the receptor, while compound 2 had only five. Compound 1 had the highest frequency of two and three H-bonds with the receptor, while one and two H-bonds for 2, indicating compound 1 was more likely to form more H-bonds with HDAC8 than compound 2, that is, the complex of HDAC8-compound 1 is more stable than HDAC8-compound 2.
Figure 10
 Figure 10. Relative frequency of the number of hydrogen bonds formed in complex (a) HDAC8-compound 1 and (b) HDAC8-compound 2 within 50 ns
Figure 10. Relative frequency of the number of hydrogen bonds formed in complex (a) HDAC8-compound 1 and (b) HDAC8-compound 2 within 50 ns3.2.5 Analysis of the covariance matrix
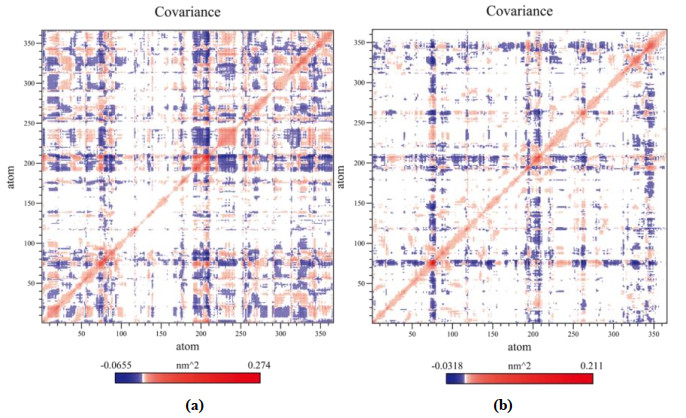
The covariance matrix was applied as a supplement for understanding the variation of residues in HDAC8. The matrix value of the white region was 0. The value of blue region was negative, representing negatively correlated motion. The value of red region was positive, showing the positively correlated variation. The covariance matrix of HDAC8 C-alpha is displayed in Fig. 11. The matrix of compound 1 has more red regions than 2. In addition, the maximum value of red region of HDAC8 with compound 1 was 0.274 and 0.211 with compound 2. These indicated that 1 prompted more positively correlated variation in HDAC8.
Figure 11
 Figure 11. Covariance matrix of (a) HDAC8 with compound 1 and (b) HDAC8 with compound 2
Figure 11. Covariance matrix of (a) HDAC8 with compound 1 and (b) HDAC8 with compound 23.3 Molecule design of new HDAC8 inhibitors
In general, the compounds containing hydroxamic acid group are mostly inhibitors of HDAC8, but their activity varies greatly due to different structures. According to the research results, a few conclusions may improve the activity of this class of compounds. First, the Cap group of the compounds with branch and long chains is advantageous to form more interactions with opening the active pocket, by which the compounds embedded in the HDAC8 like an iron nail fix their positions (as shown in Fig. 3). Second, when the ZBG and Zn ion are in the chelating positions, an O, N or F atom should be added to the part of compounds near the HIS-180 in order to form strong H-bond with the H atom of imidazole ring of HIS-180. For example, the O atom of the imide group of compound 1 forms H-bond with the H atom. This design ensures ZBG to remain in the end of the tunnel and keep chelation with Zn ion for a long time. Third, the branch chains of the Cap group can be designed to benzene rings, so that they form π-π, π-anion, π-sulfur and other interactions with the residues in pocket opening to further stabilize the position of the compounds.
4. CONCLUSION
The inhibition of HDAC8 activity by compounds containing hydroxamic acid group is mainly dependent on chelation with Zn ion. In order to maintain the interaction for a long time, the compounds stabilized in the active pocket to make sure the O atom of the hydroxamic acid group was close to the Zn ion. For the receptor, HIS-180, ASP-178, ASP-267 and GLY-140 played a significant role in stabilizing the ligand position. For ligand, the O atom of the imide group of the Cap group forming H-bond with the H atom of imidazole ring of HIS-180 played a crucial role in fixing the ligand's position. The Cap group of compound 1 is a branching structure composed of two long chains, which facilitates its interaction with the opening of active pocket and stabilizes the position of the tail of compound 1. It is like a nail in the active pocket. However, compound 2 has a short tail and no branching structure, which is unfavorable for stabilizing its location in the active pocket. Furthermore, several conclusions about the design of new high-activity HDAC8 inhibitors have been drawn: the cap group of the compounds should be designed as a long branching structure containing benzene rings. And an O, N or F atom should be added to the part of compounds near HIS-180 when the ZBG and Zn ion are in the chelating position. These conclusions have significance implications for the development of highly active HDAC8 inhibitors in the future.
-
-
[1]
Amin, S. A.; Adhikari, N.; Jha, T. Diverse classes of HDAC8 inhibitors: in search of molecular fingerprints that regulate activity. Future Med. Chem. 2018, 10, 1589−1602. doi: 10.4155/fmc-2018-0005
-
[2]
Amin, S. A.; Adhikari, N.; Jha, T. Is dual inhibition of metalloenzymes HDAC-8 and MMP-2 a potential pharmacological target to combat hematological malignancies? Pharmacol Res. 2017, 122, 8−19. doi: 10.1016/j.phrs.2017.05.002
-
[3]
Amin, S. A.; Adhikari, N.; Jha, T. Structure-activity relationships of hydroxamate-based histone deacetylase-8 inhibitors: reality behind anticancer drug discovery. Future Med. Chem. 2017, 9, 2211−2237. doi: 10.4155/fmc-2017-0130
-
[4]
Halder, A. K.; Mallick, S.; Shikha, D.; Saha, A.; Saha, K. D.; Jha, T. Design of dual MMP-2/HDAC-8 inhibitors by pharmacophore mapping, molecular docking, synthesis and biological activity. RSC Adv. 2015, 5, 72373−72386. doi: 10.1039/C5RA12606A
-
[5]
Buggy, J. J.; Sideris, M. L.; Mak, P.; Lorimer, D. D.; McIntosh, B.; Clark, J. M. Cloning and characterization of a novel human histone deacetylase, HDAC8. Biochem. J. 2000, 199−205.
-
[6]
Li, J.; Chen, S.; Cleary, R. A.; Wang, R.; Gannon, O. J.; Seto, E.; Tang, D. D. Histone deacetylase 8 regulates cortactin deacetylation and contraction in smooth muscle tissues. Am. J. Physiol. Cell Physiol. 2014, 307, 288−295. doi: 10.1152/ajpcell.00102.2014
-
[7]
Hu, E.; Chen, Z.; Fredrickson, T.; Zhu, Y.; Kirkpatrick, R.; Zhang, G. F.; Johanson, K.; Sung, C. M.; Liu, R.; Winkler, J. Cloning and characterization of a novel human class I histone deacetylase that functions as a transcription repressor. J. Biol. Chem. 2000, 275, 15254−15264. doi: 10.1074/jbc.M908988199
-
[8]
Chakrabarti, A.; Oehme, I.; Witt, O.; Oliveira, G.; Sippl, W.; Romier, C.; Pierce, R. J.; Jung, M. HDAC8: a multifaceted target for therapeutic interventions. Trends Pharmacol Sci. 2015, 36, 481−492. doi: 10.1016/j.tips.2015.04.013
-
[9]
Somoza, J. R.; Skene, R. J.; Katz, B. A.; Mol, C.; Ho, J. D.; Jennings, A. J.; Luong, C.; Arvai, A.; Buggy, J. J.; Chi, E.; Tang, J.; Sang, B. C.; Verner, E.; Wynands, R.; Leahy, E. M.; Dougan, D. R.; Snell, G.; Navre, M.; Knuth, M. W.; Swanson, R. V.; McRee, D. E.; Tari, L. W. Structural snapshots of human HDAC8 provide insights into the class I histone deacetylases. Structure 2004, 12, 1325−1334. doi: 10.1016/j.str.2004.04.012
-
[10]
Nongonierma, A. B.; Dellafiora, L.; Paolella, S.; Galaverna, G.; Cozzini, P.; FitzGerald, R. J. In silico approaches applied to the study of peptide analogs of Ile-Pro-Ile in relation to their dipeptidyl peptidase IV inhibitory properties. Front. Endocrinol. 2018, 9, 329−329. doi: 10.3389/fendo.2018.00329
-
[11]
Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; Tsuneyoshi, M. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769−774.
-
[12]
Thoma, R.; Löffler, B.; Stihle, M.; Huber, W.; Ruf, A.; Hennig, M. Structural basis of proline-specific exopeptidase activity as observed in human dipeptidyl peptidase-IV. Structure 2003, 11, 947−959. doi: 10.1016/S0969-2126(03)00160-6
-
[13]
Adhikari, N.; Amin, S. A.; Trivedi, P.; Jha, T.; Ghosh, B. HDAC3 is a potential validated target for cancer: an overview on the benzamide-based selective HDAC3 inhibitors through comparative SAR/QSAR/QAAR approaches. Eur. J. Med. Chem. 2018, 157, 1127−1142. doi: 10.1016/j.ejmech.2018.08.081
-
[14]
Wu, J.; Du, C.; Lv, Z.; Ding, C.; Cheng, J.; Xie, H.; Zhou, L.; Zheng, S. The up-regulation of histone deacetylase 8 promotes proliferation and inhibits apoptosis in hepatocellular carcinoma. Dig. Dis. Sci. 2013, 58, 3545−3553. doi: 10.1007/s10620-013-2867-7
-
[15]
Suzuki, T.; Muto, N.; Bando, M.; Itoh, Y.; Masaki, A.; Ri, M.; Ota, Y.; Nakagawa, H.; Iida, S.; Shirahige, K.; Miyata, N. Design, synthesis, and biological activity of NCC149 derivatives as histone deacetylase 8-selective inhibitors. ChemMedChem. 2014, 9, 657−664. doi: 10.1002/cmdc.201300414
-
[16]
Qi, J.; Singh, S.; Hua, W. K.; Cai, Q.; Chao, S. W.; Li, L.; Liu, H.; Ho, Y.; McDonald, T.; Lin, A.; Marcucci, G.; Bhatia, R.; Huang, W. J.; Chang, C. I.; Kuo, Y. H. HDAC8 inhibition specifically targets inv(16) acute myeloid leukemic stem cells by restoring p53 acetylation. Cell Stem. Cell. 2015, 17, 597−610. doi: 10.1016/j.stem.2015.08.004
-
[17]
Nian, H.; Bisson, W. H.; Dashwood, W. M.; Pinto, J. T.; Dashwood, R. H. Alpha-keto acid metabolites of organoselenium compounds inhibit histone deacetylase activity in human colon cancer cells. Carcinogenesis 2009, 30, 1416−1423. doi: 10.1093/carcin/bgp147
-
[18]
Hsieh, C. L.; Ma, H. P.; Su, C. M.; Chang, Y. J.; Hung, W. Y.; Ho, Y. S.; Huang, W. J.; Lin, R. K. Alterations in histone deacetylase 8 lead to cell migration and poor prognosis in breast cancer. Life Sci. 2016, 151, 7−14. doi: 10.1016/j.lfs.2016.02.092
-
[19]
Wang, Y.; Xu, P.; Yao, J.; Yang, R.; Shi, Z.; Zhu, X.; Feng, X.; Gao, S. MicroRNA-216b is down-regulated in human gastric adenocarcinoma and inhibits proliferation and cell cycle progression by targeting oncogene HDAC8. Target. Oncol. 2016, 11, 197−207. doi: 10.1007/s11523-015-0390-9
-
[20]
Oehme, I.; Deubzer, H. E.; Wegener, D.; Pickert, D.; Linke, J. P.; Hero, B.; Kopp-Schneider, A.; Westermann, F.; Ulrich, S. M.; von Deimling, A.; Fischer, M.; Witt, O. Histone deacetylase 8 in neuroblastoma tumorigenesis. Clin. Cancer Res. 2009, 15, 91−99. doi: 10.1158/1078-0432.CCR-08-0684
-
[21]
Tian, Y.; Wong, V. W.; Wong, G. L.; Yang, W.; Sun, H.; Shen, J.; Tong, J. H.; Go, M. Y.; Cheung, Y. S.; Lai, P. B.; Zhou, M.; Xu, G.; Huang, T. H.; Yu, J.; To, K. F.; Cheng, A. S.; Chan, H. L. Histone deacetylase HDAC8 promotes insulin resistance and β-catenin activation in NAFLD-associated hepatocellular carcinoma. Cancer Res. 2015, 75, 4803−4816.
-
[22]
Klebe, G.; Abraham, U.; Mietzner, T. Molecular similarity indices in a comparative analysis (CoMSIA) of drug molecules to correlate and predict their biological activity. J. Med. Chem. 1994, 37, 4130−4146. doi: 10.1021/jm00050a010
-
[23]
Doenhoff, M. J.; Cioli, D.; Utzinger, J. Praziquantel: mechanisms of action, resistance and new derivatives for schistosomiasis. Curr. Opin. Infect. Dis. 2008, 21, 659−667. doi: 10.1097/QCO.0b013e328318978f
-
[24]
Heimburg, T.; Chakrabarti, A.; Lancelot, J.; Marek, M.; Melesina, J.; Hauser, A. T.; Shaik, T. B.; Duclaud, S.; Robaa, D.; Erdmann, F.; Schmidt, M.; Romier, C.; Pierce, R. J.; Jung, M.; Sippl, W. Structure-based design and synthesis of novel inhibitors targeting HDAC8 from schistosoma mansoni for the treatment of schistosomiasis. J. Med. Chem. 2016, 59, 2423−2435. doi: 10.1021/acs.jmedchem.5b01478
-
[25]
Xia, B.; Lu, J.; Wang, R.; Yang, Z.; Zhou, X.; Huang, P. MiR-21-3p regulates influenza a virus replication by targeting histone deacetylase-8. Front Cell Infect. Microbiol. 2018, 8, 175−175. doi: 10.3389/fcimb.2018.00175
-
[26]
Meng, J.; Liu, X.; Zhang, P.; Li, D.; Xu, S.; Zhou, Q.; Guo, M.; Huai, W.; Chen, X.; Wang, Q.; Li, N.; Cao, X. Rb selectively inhibits innate IFN-β production by enhancing deacetylation of IFN-β promoter through HDAC1 and HDAC8. J. Autoimmun. 2016, 73, 42−53. doi: 10.1016/j.jaut.2016.05.012
-
[27]
Jackson, L.; Kline, A. D.; Barr, M. A.; Koch, S. De Lange syndrome: a clinical review of 310 individuals. Am. J. Med. Genet. 1993, 47, 940−946. doi: 10.1002/ajmg.1320470703
-
[28]
Ingham, O. J.; Paranal, R. M.; Smith, W. B.; Escobar, R. A.; Yueh, H.; Snyder, T.; Porco, J. A., J.; Bradner, J. E.; Beeler, A. B. Development of a potent and selective HDAC8 inhibitor. ACS Med. Chem. Lett. 2016, 7, 929−932. doi: 10.1021/acsmedchemlett.6b00239
-
[29]
Hassanzadeh, M.; Bagherzadeh, K.; Amanlou, M. A comparative study based on docking and molecular dynamics simulations over HDAC-tubulin dual inhibitors. J. Mol. Graph. Model. 2016, 70, 170−180. doi: 10.1016/j.jmgm.2016.10.007
-
[30]
Dar, K. B.; Bhat, A. H.; Amin, S.; Hamid, R.; Anees, S.; Anjum, S.; Reshi, B. A.; Zargar, M. A.; Masood, A.; Ganie, S. A. Modern computational strategies for designing drugs to curb human diseases: a prospect. Curr. Top Med. Chem. 2018, 18, 2702−2719.
-
[31]
Zhou, H.; Wang, C.; Deng, T.; Tao, R.; Li, W. Novel urushiol derivatives as HDAC8 inhibitors: rational design, virtual screening, molecular docking and molecular dynamics studies. J. Biomol. Struct. Dyn. 2018, 36, 1966−1978. doi: 10.1080/07391102.2017.1344568
-
[32]
Uba, A. I.; Weako, J.; Keskin, Ö.; Gürsoy, A.; Yelekçi, K. Examining the stability of binding modes of the co-crystallized inhibitors of human HDAC8 by molecular dynamics simulation. J. Biomol. Struct. Dyn. 2020, 38, 1751−1760.
-
[33]
Zhang, Y.; Feng, J.; Jia, Y.; Wang, X.; Zhang, L.; Liu, C.; Fang, H.; Xu, W. Development of tetrahydroisoquinoline-based hydroxamic acid derivatives: potent histone deacetylase inhibitors with marked in vitro and in vivo antitumor activities. J. Med. Chem. 2011, 54, 2823−2838. doi: 10.1021/jm101605z
-
[34]
Zhang, Y. J.; Feng, J. H.; Liu, C. X.; Fang, H.; Xu, W. F. Design, synthesis and biological evaluation of tyrosine-based hydroxamic acid analogs as novel histone deacetylases (HDACs) inhibitors. Bioorg. Med. Chem. 2011, 19, 4437−4444. doi: 10.1016/j.bmc.2011.06.046
-
[35]
Zhang, Y. J.; Feng, J. H.; Liu, C. X.; Zhang, L.; Jiao, J.; Fang, H.; Su, L.; Zhang, X. P.; Zhang, J.; Li, M. Y.; Wang, B. H.; Xu, W. F. Design, synthesis and preliminary activity assay of 1, 2, 3, 4-tetrahydroisoquinoline-3-carboxylic acid derivatives as novel histone deacetylases (HDACs) inhibitors. Bioorg. Med. Chem. Lett. 2010, 18, 1761−1772. doi: 10.1016/j.bmc.2010.01.060
-
[36]
Pidugu, V. R.; Yarla, N. S.; Pedada, S. R.; Kalle, A. M.; Satya, A. K. Design and synthesis of novel HDAC8 inhibitory 2, 5-disubstituted-1, 3, 4-oxadiazoles containing glycine and alanine hybrids with anti cancer activity. Bioorg. Med. Chem. 2016, 24, 5611−5617. doi: 10.1016/j.bmc.2016.09.022
-
[37]
Qi, C. Y.; Zhang, R.; Liu, F. Z.; Zheng, T.; Wu, W. J. Molecular mechanism of interactions between inhibitory tripeptide GEF and angiotensin-converting enzyme in aqueous solutions by molecular dynamic simulations. J. Mol. Liq. 2018, 249, 389−396. doi: 10.1016/j.molliq.2017.11.033
-
[38]
Yan, W. L.; Lin, G. M.; Zhang, R.; Liang, Z.; Wu, W. J. Studies on the bioactivities and molecular mechanism of antioxidant peptides by 3D-QSAR, in vitro evaluation and molecular dynamic simulations. Food Funct. 2020, 11, 3043−3052. doi: 10.1039/C9FO03018B
-
[39]
Tabackman, A. A.; Frankson, R.; Marsan, E. S.; Perry, K.; Cole, K. E. Structure of 'linkerless' hydroxamic acid inhibitor-HDAC8 complex confirms the formation of an isoform-specific subpocket. J. Struct. Biol. 2016, 195, 373−378. doi: 10.1016/j.jsb.2016.06.023
-
[40]
Lombardi, P. M.; Cole, K. E.; Dowling, D. P.; Christianson, D. W. Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 2011, 21, 735−743. doi: 10.1016/j.sbi.2011.08.004
-
[41]
Deschamps, N.; Simões-Pires, C. A.; Carrupt, P. A.; Nurisso, A. How the flexibility of human histone deacetylases influences ligand binding: an overview. Drug Discov. Today 2015, 20, 736−742. doi: 10.1016/j.drudis.2015.01.004
-
[42]
Nechay, M. R.; Gallup, N. M.; Morgenstern, A.; Smith, Q. A.; Eberhart, M. E.; Alexandrova, A. N. Histone deacetylase 8: characterization of physiological divalent metal catalysis. J. Phys. Chem. B 2016, 120, 5884−5895. doi: 10.1021/acs.jpcb.6b00997
-
[1]
-
Figure 2 (a) Interactions between the active site of HDAC8 (color: green) and compound 1 (color: wheat). Metal chelating bond, H-bonds, π-π T-shaped and π-anion are depicted as cyan, yellow, red and blue dotted lines, respectively. (b) The Zn ion is shown as dark grey sphere. Interactions between the active site of HDAC8 (color: green) and compound 2 (color: wheat). Metal chelating bonds, H-bonds, π-π T-shaped and π-sulfur are depicted as cyan, yellow, red and orange dotted lines, respectively. The Zn ion is shown as dark grey sphere
Figure 3 (a) Conformation of compound 1 bound to the active site. (b) Conformation of compound 2 bound to the active site
Figure 4 Hydrophobic interaction of complex system (a) HDAC8-compound 1 and (b) HDAC8-compound 2
Figure 6 Fluctuation of binding free energy of complex (a) HDAC8-compound 1 and (b) HDAC8-compound 2
Figure 9 Distance between Zn and the O atom on the carbonyl group of hydroxamic acid group
Figure 10 Relative frequency of the number of hydrogen bonds formed in complex (a) HDAC8-compound 1 and (b) HDAC8-compound 2 within 50 ns
Figure 11 Covariance matrix of (a) HDAC8 with compound 1 and (b) HDAC8 with compound 2
Table 1. Binding Energy Contributions between HDAC8 Residues and Compounds 1 and 2
Residues and ions of HDAC8 Binding energy of HDAC8 and Compound 1 (kJ/mol) Residues and ions of HDAC8 Binding energy of HDAC8 and Compound 2 (kJ/mol) ZN-388 –197.506 ZN-388 48.927 PHE-208 –63.600 ASP-101 –52.187 HIS-180 –16.439 ASP-272 –34.813 MET-274 –14.043 ASP-267 –29.639 PHE-207 –11.505 ASP-178 –23.193 PHE-152 –10.415 ASP-29 –21.349 TRP-141 –9.116 GLU-148 –20.505 TYR-306 –6.959 ASP-183 –20.450 ASP-101 –6.828 ASP-233 –19.657 LYS-202 –5.891 ASP-87 –18.916 SER-150 –5.427 ASP-157 –18.713  下载: 导出CSV
下载: 导出CSV
-












 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 1726
- HTML全文浏览量: 14
 下载:
下载:
