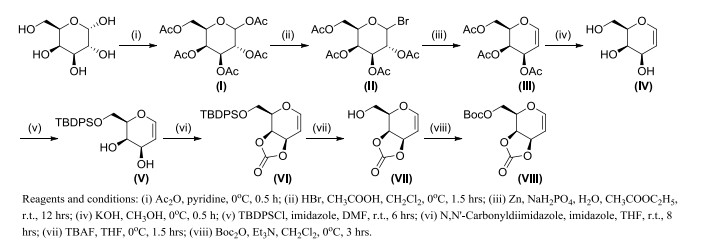
Scheme 1.
Synthetic routes for the title intermediates VIII
Synthesis and Crystal Structure of tert-Butyl(((2R, 3R, 6R)-3-hydroxy-6-(nitromethyl)-3, 6-dihydro-2H-pyran-2-yl)methyl)carbonate
Yong-Kui FENG , Hua-Lan SU , Billy Joel MUKULA OTUKOL , Joel Billy , Xue-Qing ZHANG , Hui YAO , Nian-Yu HUANG

C-glycosides are an important part of carbohydrate chemistry because they have been embedded in a variety of natural products with significant biological importance[1, 2]. Besides their potential bioactivity, C-glycosides are more stable than native N- or O-glycosylated residues, since N- or O-glycosidic bonds can be cleaved enzymatically under physiological conditions[3-5]. C-glycosides can behave like comparable conformations of O- and N-glycosides. Consequently, they can be beneficial as substantial mimics of biologically active natural O-glycosides and thus can be employed as potential therapeutic agents[6, 7]. For example, C-glycoside drugs could be used as the inhibitors of sodium glucose co-transporter 2 (SGLT2)[8], including dapagliflozin[9], canagliflozin[10] and empagliflozin[11], which have received considerable attention for the treatment of type II diabetes mellitus.
Chemical C-glycosylation has been well developed in recent years due to the extremely diverse structures of naturally existing C-glycosides. By activating the anomeric center with the exploitation of Lewis acid catalysts, C-nucleophiles have been utilized predominantly for the formulation of C-glycosidic linkages[12, 13]. Although Ferrier glycosylation[14], Heck reaction[15] and Tsuji-Trost reaction[16] have been applied successfully in C-glycosylation with a catalytic amount of transition-metal catalysts, most of these reactions need to be carried out at high temperature or aided by additives. Therefore, many efforts have been focused on developing milder, more efficient and highly stereoselective alternatives such as transition metal-catalyzed C-glycosylation of unsaturated glycoside donors. Transition metal-catalyzed cross-coupling reactions are being developed as a diverse approach for the synthesis of naturally abundant C-glycosides and have emerged as a powerful method for the construction of C-glycosides[17-19]. Inspired by the above researches, we designed and synthesized the unsaturated Boc-protected 3, 4-O-carbonate glycal donor (VIII, Scheme 1), then treated it to C-glycoside via transition metal-catalyzed cross-coupling reactions under mild conditions in this work (Scheme 2).

Most reagents were obtained from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China), and were used without further purification. Solvents were dried and purified using standard techniques. D-Galactose was purchased from J & K Chemical Co., Ltd., China. Reactions were monitored by thin layer chromatography on silica gel GF254 pre-coated plates. 1D and 2D nuclear magnetic resonance (NMR) spectra were recorded at a Bruker UltrashiedTM 400 MHz Plus spectrometer. Chemical shifts (δ) were reported in ppm, using residual solvent as an internal standard. Melting points were tested with an uncorrected X-4 digital melting point apparatus. High-resolution electrospray ionization mass spectra (HR-ESI-MS) were obtained using a Waters Q-TOF premierTM mass spectrometer. Characterization data for known compounds were checked in comparison with literature for consistency and not presented in this report.
D-Galactose (10.0 g, 55.6 mmol) was dissolved in anhydrous pyridine (100 mL), and acetic anhydride (30 mL) was slowly added dropwise to the mixture at 0 oC. After stirring at room temperature for 30 minutes, the reaction was quenched with H2O (200 mL) and extracted with ethyl acetate (40 mL × 3). The extracts were combined, dried with Na2SO4, filtered, and evaporated to dryness to give the crude beta-D-galactose pentaacetate (I), which could be used directly in the next step without further purification.
The crude beta-D-galactose pentaacetate (I, 10.0 g, 25.6 mmol) was dissolved in CH2Cl2 (200 mL), and the solution of HBr/AcOH was slowly added at 0 oC. The mixture was stirred at room temperature for 1.5 hours and thin-layer chromatography (TLC) was used to monitor the process until completion. The mixture was quenched with H2O (200 mL), neutralized with saturated NaHCO3 solution, and extracted with CH2Cl2 (50 mL × 3). The combined organic extracts were dried with Na2SO4 and filtered, and evaporated to dryness to give the oily product, which was purified by silica gel column chromatography (eluent: petroleum ether/ethyl acetate = 15/1, v/v) to give the 2, 3, 4, 6-tetra-O-acetyl-alpha-D-galactopyranosyl bromide (II) as colorless syrup[20] (9.5 g, yield 90%). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.61 (d, J = 4.0 Hz, 1H), 5.56 (t, J = 9.7 Hz, 1H), 5.17 (t, J = 9.8 Hz, 1H), 4.84 (dd, J = 10.0, 4.1 Hz, 1H), 4.39~4.25 (m, 2H), 4.19~4.08 (m, 1H), 2.11 (s, 3H), 2.10 (s, 3H), 2.06 (s, 3H), 2.04 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.4, 169.8, 169.7, 169.4, 86.5, 72.1, 70.5, 70.1, 67.1, 60.9, 20.6, 20.6, 20.5, 20.5.
The suspension mixture containing compound II (9.5 g, 24 mmol) and zinc powder (30.0 g, 461 mmol) in saturated NaH2PO4 aqueous solution (100 mL) and ethyl acetate (100 mL) was vigorously stirred at room temperature for 12 hours and TLC was used to monitor the reaction until completion. The solid was filtered and organic layer was separated. The aqueous phase was extracted with ethyl acetate (40 mL × 3). The organic extracts were combined, dried with Na2SO4 and filtered, and evaporated to dryness to give the crude product, which was purified by silicagel flash chromatography with a gradient solvent system (eluent: petroleum ether/ethyl acetate = 10/1, v/v) to obtain the (2R, 3R, 4R)-2-(acetoxymethyl)-3, 4-dihydro-2H-pyran-3, 4-diyl diacetate (III) as colorless syrup[21] (5.8 g, yield 92%). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.48 (dd, J = 6.3, 1.4 Hz, 1H), 5.63~5.50 (m, 1H), 5.44 (d, J = 4.5 Hz, 1H), 4.73~4.76 (m, 1H), 4.36~4.19 (m, 3H), 2.15 (s, 3H), 2.10 (s, 3H), 2.04 (s, 3H). 13C NMR (100 MHz, CDCl3) δ (ppm): 170.6, 170.3, 170.1, 145.4, 98.8, 72.7, 63.8, 63.7, 61.9, 20.8, 20.8, 20.7.
Compound III (5.8 g, 21.3 mmol) and KOH (2.0 g, 36 mmol) were dissolved in methanol (50 mL) and stirred at 0 oC for 30 minutes. TLC was used to monitor the process until the reaction was complete. After removal of the solvent, the residue was purified by silica gel flash chromatography (eluent: ethyl acetate/methanol = 50/1, v/v) to get the D-galactal (IV) as white solids[21] (4.0 g, yield 83%, m.p. 99~100 oC). 1H NMR (400 MHz, DMSO-d6) δ (ppm): 6.26 (dd, J = 6.2, 1.9 Hz, 1H), 4.74 (t, J = 5.6 Hz, 1H), 4.64 (d, J = 6.7 Hz, 1H), 4.48 (dt, J = 6.2, 2.0 Hz, 1H), 4.39 (d, J = 4.8 Hz, 1H), 4.22~4.17 (m, 1H), 3.79~3.67 (m, 2H), 3.63~3.52 (m, 2H).13C NMR (100 MHz, DMSO-d6) δ (ppm): 143.5, 104.1, 77.9, 64.6, 63.8, 60.7.
To a mixture of compound IV (4.0 g, 27.4 mmol), tert-butyldiphenylchlorosilane (TBDPSCl, 8.46 g, 30.1 mmol) and imidazole (4.2 g, 61 mmol) were dissolved in N, N΄-dimethylformamide (DMF, 60 mL), and stirred at room temperature for 6 hours until the completion of the reaction monitored by TLC. The reaction was quenched by water and crude product was extracted by ethyl acetate. After removing the solvent, pure (2R, 3R, 4R)-2-(((tert-Butyldiphenylsilyl)oxy)methyl)-3, 4-dihydro-2H-pyran-3, 4-diol (V) was obtained by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 1/1, v/v) as colorless syrup[22] (9.2 g, yield 84%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.83~7.62 (m, 4H), 7.55~7.34 (m, 6H), 6.41 (dd, J = 6.2, 1.5 Hz, 1H), 4.75 (dt, J = 6.2, 1.9 Hz, 1H), 4.46~4.29 (m, 1H), 4.17 (t, J = 4.8 Hz, 1H), 4.01 (dd, J = 11.9, 6.9 Hz, 1H), 3.96~3.88 (m, 2H), 2.99 (d, J = 5.3 Hz, 1H), 2.60 (d, J = 10.2 Hz, 1H), 1.09 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 144.4, 135.6, 135.5, 132.6, 132.4, 130.0, 127.8, 127.8, 103.4, 65.8, 64.4, 63.8, 26.7, 19.1.
Compound V (8.7 g, 22.8 mmol), N, N΄-carbonyldiimidazole (7.3 g, 45.6 mmol) and catalytic amount of imidazole (10 mg) were dissolved in anhydrous tetrahydrofuran (THF, 100 mL) and stirred at room temperature for 8 hours by TLC monitoring until all substrate was fully consumed. The mixture was quenched by ice water (100 mL), and crude product was extracted by ethyl acetate (60 mL × 3). After separating the organic layer and removal of the solvent, the oily crude was purified by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 8/1, v/v) to give 1, 5-anhydro-6-O-(tert-butyldiphenylsilyl)-3, 4-O-carbonate-2-deoxy-D-lyxo-hex-1-enopyranose (VI) as colorless syrup[22] (4.5 g, yield 83%). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.76~7.68 (m, 4H), 7.54~7.43 (m, 6H), 6.68 (d, J = 6.3 Hz, 1H), 5.25 (dd, J = 7.7, 3.1 Hz, 1H), 5.10 (d, J = 7.7 Hz, 1H), 4.99 (ddd, J = 6.2, 3.1, 1.1 Hz, 1H), 4.08~3.97 (m, 3H), 1.13 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 154.2, 149.1, 135.5, 135.5, 134.8, 132.7, 132.5, 130.1, 130.0, 127.9, 127.9, 127.7, 98.0, 73.8, 72.9, 68.9, 61.7, 26.8, 26.6, 19.2.
Compound VI (1.5 g, 3.6 mmol) and tetrabutylammonium fluoride hydrate (TBAF, 1.3 g, 3.9 mmol) were dissolved in anhydrous THF (30 mL). The reaction mixture was stirred at 0 oC for 1.5 hours. TLC was employed to monitor the reaction until it was complete. The subsequent mixture was thereafter concentrated to obtain a crude product which was purified by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 4/1, v/v) to give the (3aR, 4R, 7aR)-4-(hydroxymethyl)-3a, 7a-dihydro-4H-[1, 3]dioxolo[4, 5-c]-pyran-2-one (VII) as colorless syrup[23] (623 mg, yield 86%). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.74 (d, J = 6.3 Hz, 1H), 5.24 (dd, J = 7.7, 3.2 Hz, 1H), 5.01 (ddd, J = 6.3, 3.2, 1.2 Hz, 1H), 4.98~4.91 (m, 1H), 4.10~3.99 (m, 2H), 3.93 (d, J = 9.6 Hz, 1H), 2.24 (s, 1H). 13C NMR (100 MHz, CDCl3) δ (ppm): 154.0, 149.1, 98.1, 73.9, 73.1, 69.0, 61.5, 29.7.
The mixture of VII (600 mg, 3.4 mmol), Et3N (1.06 mL, 10.4 mmol) and di-tert-butyldicarbonate (912 mg, 4.2 mmol) was dissolved in anhydrous CH2Cl2 (30 mL) and stirred at 0 oC for 3 hours. The reaction was monitored with TLC analysis until the completion. The solvent was removed under reduced pressure to afford a crude product, which was purified by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 8/1, v/v) to get the tert-butyl(((3aR, 4R, 7aR)-2-oxo-3a, 7a-dihydro-4H-[1, 3]dioxolo-[4, 5-c]pyran-4-yl)methyl)carbonate (VIII) as yellow syrup[22] (706 mg, yield 87%). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.71 (d, J = 6.3 Hz, 1H), 5.20 (dd, J = 7.7, 3.2 Hz, 1H), 5.00 (ddd, J = 6.3, 3.2, 1.1 Hz, 1H), 4.91 (d, J = 7.7 Hz, 1H), 4.42 (dd, J = 11.6, 7.2 Hz, 1H), 4.34 (dd, J = 11.6, 5.6 Hz, 1H), 4.21~4.12 (m, 1H), 1.50 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 149.1, 98.1, 83.3, 72.9, 71.5, 68.6, 64.6, 29.7, 27.7.
To the solution of CH2Cl2 (5 mL) of D-galactal carbonate (VIII, 272 mg, 1.0 mmol) and nitromethane (122 mg, 0.2 mmol) were added palladium acetylacetonate (2.3 mg, 0.0075 mmol) and 1, 4-bis(diphenylphosphino)butane (DPPB, 2.2 mg, 0.005 mmol) under nitrogen atmosphere, and the reaction mixture was stirred at room temperature for 24 hours until the completion of reaction (TLC monitoring). The reaction was quenched by ice water (10 mL), and crude product was extracted by ethyl acetate (10 mL × 3). After removing the solvent, the crude product was purified by silica gel flash chromatography (eluent: petroleum ether/ethyl acetate = 10/1, v/v) to give the target product (IX) as white solids (257 mg, yield 91%. m.p. 111~112 oC). 1H NMR (400 MHz, CDCl3) δ (ppm): 6.28 (ddd, J = 10.1, 5.7, 2.2 Hz, 1H), 5.92 (dd, J = 10.4, 1.2 Hz, 1H), 4.91~4.84 (m, 1H), 4.59~4.55(m, 2H), 4.37 (dd, J = 11.6, 6.0 Hz, 1H), 4.29 (dd, J = 11.6, 7.2 Hz, 1H), 4.02~3.95 (m, 1H), 3.86~3.82 (m, 1H), 2.04 (d, J = 9.9 Hz, 1H), 1.53 (s, 9H). 13C NMR (100 MHz, CDCl3) δ (ppm): 153.4, 129.8, 128.2, 82.7, 77.9, 75.8, 71.9, 65.7, 61.7, 27.7. HRMS calcd. for C12H19NO7Na [M+Na]+: 289.1162. Found 289.1169; [α]D25 = –71.803. (c = 1.0, CHCl3).
Compound IX crystallized after slow evaporation from a saturated chloroform solution as colorless blocks with dimensions of 0.14mm × 0.13mm × 0.11mm in the monoclinic system. Diffraction data of the single crystal were obtained at 100 K from a Bruker SMART APEX-II CCD diffractometer with graphite-monochromated CuKα radiation (λ = 1.54184 Å) using the Bruker Collect software. A total of 5644 reflections were collected in the range of 2.39≤θ≤73.69º by using an ω-scan mode, of which 2875 were unique with Rint = 0.0163 and 2857 were observed with I > 2σ(I). After the initial corrections and data reduction, intensities of reflections were used to solve (by direct methods) and refine the structures (on F2) using the WINGX program. A weighting scheme based upon P = (Fo2 + 2Fc2)/3 was employed. All the hydrogen atoms were located from difference maps and included in the refinements as riding. Empirical absorption corrections were applied. The structures were solved by direct methods using SHELXL-97 programs[23]. All of the non-hydrogen atoms were located from difference Fourier maps, and then refined anisotro-pically with SHELXL-97 via a full-matrix least-square procedure[23]. The final R = 0.0294, wR = 0.0752, (Δ/σ)max = 0.000, S = 1.048, (Δρ)max = 0.138 and (Δρ)min = –0.253 e/Å3. The Flack parameter was –0.01(6).
The α-glucosidase inhibitory activity was performed according to a published method[24, 25]. 3 mM p-nitro-phenyl-α-D-glucopyranoside (20 μL) and 0.2 U/mL α-glucosidase (20 μL) in 0.01 M phosphate buffer (pH = 7.0) were added to the sample solution and dissolved in dimethyl sulfoxide (DMSO, 10 μL) to start the reaction. Each reaction was carried out at 37 ºC for 30 min and stopped by adding 0.1 M Na2CO3 (150 μL). The absorbance was recorded at 410 nm. All the samples were tested in triplicate, and the IC50 values were calculated from the dose-inhibition curve plotting using six different sample concentrations. 1-Deoxynojirimycin[26], a known α-glucosidase inhibitor, was used as a positive control.
The key intermediate D-galactal carbonate (VIII) was synthesized from D-galactose through an eight-step reaction with a total yield of 35% (Scheme 1). The glycosylation reaction was conducted by the palladium catalyst in the presence of phosphorus ligands (P-ligands) including 4, 5-bis(diphnyl-phos-phino)-9, 9-dimethylxan-thene (xantphos), 1, 4-bis(diphenylphosphino)-butane (DPPB), 1, 1΄-bis(diphenylphosphino)ferrocene (DPPF), 2-di-tert-butylphosphino-2΄, 4΄, 6΄-triiso-propylbiphenyl (tBuXPhos) and tricyclohexyl-phosphine (P(Cy)3). By screening different palladium catalysts and P-ligands (Scheme 2), β-C-pyranogalactoside (IX) was obtained with a satisfactory yield of 91% under optimal conditions using 2.5 mol% Pd(acac)2 catalyst, and 5 mol% DPPB in dichloromethane at room temperature (Table 1).
 DownLoad:
CSV
DownLoad:
CSV
| Entrya | Catalyst | P-Ligand | Solvent | Yieldb |
| 1 | Pd2(dba)3 | Xantphos | THF | 40% |
| 2 | Pd(OAc)2 | Xantphos | THF | 10% |
| 3 | White catalyst | Xantphos | THF | - |
| 4 | Pd(PPh3)4 | Xantphos | THF | - |
| 5 | PdCl2 | Xantphos | THF | - |
| 6 | Pd(acac)2 | Xantphos | THF | 60% |
| 7 | Pd(acac)2 | DPPB | THF | 86% |
| 8 | Pd(acac)2 | DPPF | THF | 74% |
| 9 | Pd(acac)2 | tBuXPhos | THF | 66% |
| 10 | Pd(acac)2 | P(Cy)3 | THF | 57% |
| 11 | Pd(acac)2 | DPPB | CH2Cl2 | 91% |
| 12 | Pd(acac)2 | DPPB | CH3CN | 88% |
| 13 | Pd(acac)2 | DPPB | Toluene | 76% |
| aUnless otherwise specified, all reactions were carried out with 0.1 mmol of IX, 0.2 mmol of nitromethane, 2.5 mol% Pd catalyst and 5 mol% P-ligand in 2 mL solvent and N2 atmosphere at room temperature. bIsolated yield, N.R. = No reaction. | ||||
All the intermediates (II~VIII) were characterized by NMR, and their data obtained were found to be consistent with that of other literatures. In the 1H NMR spectrum, the protons of the carbon-carbon double bond for the title compound (IX) were located at 6.28 and 5.92 ppm, and the hydrogen atoms of tert-butyl group were found as singlet at 1.53 ppm. Two sets of carbon signals for the methylene group appeared at 65.76 and 61.71 ppm in its 13C NMR and DEPT-135 spectra. The adduction [M + Na]+ in the HR-ESI-MS spectrum could be also observed for the title compound.
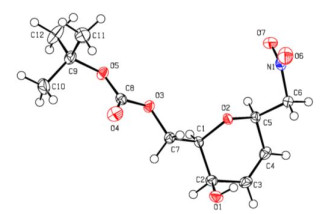
The selected bond lengths, bond angles and torsion angles are listed in Table 2, and all the bond lengths of C–C, C=C and C=O were in accordance with the standard compilations and the literature[27]. The bond angles containing unsaturated bonds in C(3)–C(4)–C(5), O(4)–C(8)–O(3) and O(6)–N(1)– O(7) range from 112° to 126°. The torsion angles of sugar chain O(2)–C(1)–C(2)–C(3) and O(2)–C(5)–C(4)–C(3) were equal to –49.34(16)° and 11.5(2)°, respectively. The aliphatic 6-membered ring O(2)–C(1)–C(2)–C(3)–C(4)–C(5) showed a twist-boat form (Φ = 217.6139°, θ = 128.53°, puckering amplitude (Q) = 0.4921°).
DownLoad:
CSV
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| C(1)–O(2) | 1.4383(17) | C(5)–C(6) | 1.518(2) | O(3)–C(7) | 1.4493(17) | ||
| O(2)–C(5) | 1.4233(17) | N(1)–C(6) | 1.493(2) | O(4)–C(8) | 1.1994(19) | ||
| C(5)–C(4) | 1.500(2) | O(6)–N(1) | 1.215(2) | O(5)–C(8) | 1.3280(19) | ||
| C(4)–C(3) | 1.325(2) | O(7)–N(1) | 1.2335(19) | O(1)–C(2) | 1.4305(19) | ||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| C(5)–O(2)–C(1) | 112.11(11) | O(6)–N(1)–O(7) | 124.16(15) | O(2)–C(1)–C(7)–O(3) | 83.57(13) | ||
| C(3)–C(4)–C(5) | 122.03(14) | O(4)–C(8)–O(3) | 125.22(15) | O(2)–C(1)–C(2)–O(1) | 74.93(15) | ||
| C(3)–C(4)–C(5) | 122.03(14) | C(8)–O(5)–C(9) | 121.05(12) | O(2)–C(1)–C(2)–C(3) | –49.34(16) | ||
| C(7)–C(1)–C(2) | 111.21(12) | C(11)–C(9)–C(10) | 112.40(15) | O(2)–C(5)–C(4)–C(3) | 11.5(2) |
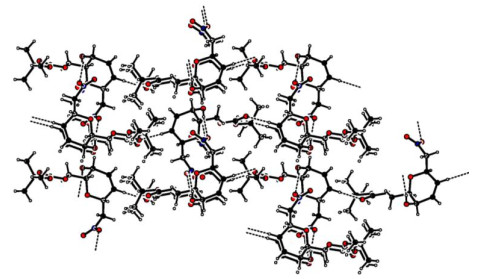
As listed in Table 3, intermolecular and intramolecular O–H⋅⋅⋅O and C–H⋅⋅⋅O interactions linked the molecules into a two-dimensional infinite plane running along the c axis, which helped to stabilize the crystal structure (Fig. 2).
DownLoad:
CSV
| D–H⋅⋅⋅A | d(D–H) | d(H⋅⋅⋅A) | d(D⋅⋅⋅A) | < (DHA) |
| O(1)–H(1)⋅⋅⋅O(7)(a) | 0.82 | 2.08 | 2.8904(1) | 172 |
| C(4)–H(4)⋅⋅⋅O(4)(b) | 0.93 | 2.37 | 3.2683(1) | 163 |
| C(10)–H(10C)⋅⋅⋅O(4) | 0.96 | 2.37 | 2.9117(1) | 115 |
| Symmetry codes: (a) –1/2 + x, –1/2 – y, –z; (b) 1/2 – x, –y, –1/2 + z | ||||
C-glycoside derivatives have been reported to possess α-glucosidase inhibitory activity[9-11] that is a main target enzyme in the prevention and treatment of type 2 diabetes[28]. As our continuous interest in search of natural products-derived biological small molecules[29-32], the β-C-pyranogalactoside (IX) and its synthetic intermediates (IV-VIII) were evaluated for α-glucosidase inhibitory activity (Table 4). Compared with the positive control of 1-deoxynojirimycin (IC50: 0.29 μg/mL), all the synthetic intermediates (IV-VIII) exhibited poor inhibitory effect against α-glucosidase (IC50 > 200 μg/mL), and the target C-glycoside (IX) just showed moderate inhibitory activity with the IC50 value of 89.86 μg/mL. Further exploration of the substrate scope for this reaction and structural modification of the C-glycosides were underway in our research group.
DownLoad:
CSV
| Compound | IC50 (μg/mL) | Compound | IC50 (μg/mL) | |
| IV | > 200 | VIII | > 200 | |
| V | > 200 | IX | 89.86 | |
| VI | > 200 | 1-Deoxynojirimycina | 0.29 | |
| VII | > 200 | DMSOb | - | |
| aPositive control, bblank test. | ||||
In summary, a β-C-pyranogalactoside (IX) was prepared by an efficient and highly stereoselective approach from D-galactose, and its absolute configuration was confirmed with a Flack parameter of –0.01(6) by X-ray crystallography. The in vitro α-glucosidase inhibitory activity evaluation indicated that C-pyranoside showed better inhibitory effect than the synthetic D-galactal intermediates, which encouraged us to further investigate the reaction with the aim of systematically assessing the biological activity for the C-glycosides.
Liao, H. Z.; Ma, J. M.; Yao, H.; Liu, X. W. Recent progress of C-glycosylation methods in the total synthesis of natural products and pharmaceuticals. Org. Biomol. Chem. 2018, 16, 1791−1806. doi: 10.1039/C8OB00032H
Yang, Y.; Yu, B. Recent advances in the chemical synthesis of C-glycosides. Chem. Rev. 2017, 117, 12281−12356. doi: 10.1021/acs.chemrev.7b00234
Wei, B.; Wang, Y. K.; Qiu, W. H.; Wang, S. J.; Wu, Y. H.; Xu, X. W.; Wang, H. Discovery and mechanism of intestinal bacteria in enzymatic cleavage of C−C glycosidic bonds. Appl. Microbiol. Biotechnol. 2020, 104, 1883−1890. doi: 10.1007/s00253-019-10333-z
Drohat, A. C.; Maiti, A. Mechanisms for enzymatic cleavage of the N-glycosidic bond in DNA. Org. Biomol. Chem. 2014, 12, 8367−8378. doi: 10.1039/C4OB01063A
Wolfenden, R.; Lu, X.; Young, G. Spontaneous hydrolysis of glycosides. J. Am. Chem. Soc. 1998, 120, 6814−6815. doi: 10.1021/ja9813055
Yang, G.; Schmieg, J.; Tsuji, M.; Franck, R. W. The C-glycoside analogue of the immunostimulant α-galactosylceramide (KRN7000): synthesis and striking enhancement of activity. Angew. Chem. Int. Ed. 2004, 43, 3818‒3822. doi: 10.1002/anie.200454215
Kitamura, K.; Ando, Y.; Matsumoto, T.; Suzuki, K. Total synthesis of aryl C-glycoside natural products: strategies and tactics. Chem. Rev. 2018, 118, 1495‒1598. doi: 10.1021/acs.chemrev.7b00380
Rieg, T.; Vallon, V. Development of SGLT1 and SGLT2 inhibitors. Diabetologia 2018, 61, 2079‒2086. doi: 10.1007/s00125-018-4654-7
Dhillon, S. Dapagliflozin: a review in type 2 diabetes. Drugs 2019, 79, 1135‒1146. doi: 10.1007/s40265-019-01148-3
Deeks, E. D.; Scheen, A. J. Canagliflozin: a review in type 2 diabetes. Drugs 2017, 77, 1577‒1592. doi: 10.1007/s40265-017-0801-6
Levine, M. J. Empagliflozin for type 2 diabetes mellitus: an overview of phase 3 clinical trials. Curr. Diabetes Rev. 2017, 13, 405‒423.
Sakamoto, K.; Nagai, M.; Ebe, Y.; Yorimitsu, H.; Nishimura, T. Iridium-promoted deoxyglycoside synthesis: stereoselectivity and mechanistic insight. ACS Catal. 2019, 9, 1347–1352. doi: 10.1021/acscatal.8b04686
Zhu, F.; Rourke, M. J.; Yang, T.; Rodriguez, J.; Walczak, M. A. Highly stereospecific cross-coupling reactions of anomeric stannanes for the synthesis of C-aryl glycosides. J. Am. Chem. Soc. 2016, 138, 12049–12052. doi: 10.1021/jacs.6b07891
Wang, J.; Deng, C.; Zhang, Q.; Chai, Y. Tuning the chemoselectivity of silyl protected rhamnals by temperature and bronsted acidity: kinetically controlled 1, 2-addition vs thermodynamically controlled Ferrier rearrangement. Org. Lett. 2019, 21, 1103–1107. doi: 10.1021/acs.orglett.9b00009
Mabit, T.; Siard, A.; Legros, F.; Guillarme, S.; Martel, A.; Lebreton, J.; Carreaux, F.; Dujardin, G.; Collet, S. Stereospecific C-glycosylation by Mizoroki-Heck reaction: a powerful and easy-to-set-up synthetic tool to access alpha- and beta-aryl-C-glycosides. Chem. Eur. J. 2018, 24, 14069–14074. doi: 10.1002/chem.201803674
Leng, W. L.; Liao, H. Z.; Yao, H.; Ang, Z. E.; Xiang, S. H.; Liu, X. W. Palladium-catalyzed decarboxylative allylation/Wittig reaction: substrate-controlled synthesis of C-vinyl glycosides. Org. Lett. 2017, 19, 416–419. doi: 10.1021/acs.orglett.6b03697
Li, W.; Yu, B. Gold-catalyzed glycosylation in the synthesis of complex carbohydrate-containing natural products. Chem. Soc. Rev. 2018, 47, 7954–7984. doi: 10.1039/C8CS00209F
Ye, W.; Stevens, C. M.; Wen, P.; Simmons, C. J.; Tang, W. Mild Cu(OTf)2-mediated C-glycosylation with chelation-assisted picolinate as a leaving group. J. Org. Chem. 2020, 85, 16218–16225. doi: 10.1021/acs.joc.0c01041
Zhu, F.; Rodriguez, J.; Yang, T.; Kevlishvili, I.; Miller, E.; Yi, D.; O'Neill, S.; Rourke, M. J.; Liu, P.; Walczak, M. A. Glycosyl cross-coupling of anomeric nucleophiles: scope, mechanism, and applications in the synthesis of aryl C-glycosides. J. Am. Chem. Soc. 2017, 139, 17908–17922. doi: 10.1021/jacs.7b08707
Liu, S. X.; Tsai, Y. T.; Lin, Y. T.; Li, J. Y.; Chang, C. C. Design and synthesis of trivalent Tn glycoconjugate polymers by nitroxide-mediated polymerization. Tetrahedron 2019, 75, 1307–1311.
Moore, P. W.; Schuster, J. K.; Hewitt, R. J.; Stone, M. R. L.; Harvey, J. E. Divergent synthesis of 2-C-branched pyranosides and oxepines from 1, 2-gem-dibromocyclopropyl carbohydrates. Tetrahedron 2014, 70, 7032–7043. doi: 10.1016/j.tet.2014.06.069
Dai, Y. W.; Zheng, J. F.; Zhang, Q. General strategy for stereoselective synthesis of β-N-glycosyl sulfonamides viapalladium-catalyzed glycosylation. Org. Lett. 2018, 20, 3923–3927. doi: 10.1021/acs.orglett.8b01506
Sheldrick, G. M. SHELXL 97, Program for the Refinement of Crystal Structure. University of Göttingen, Germany 1997.
Ma, Y. Y.; Zhao, D. G.; Zhang, R.; He, X.; Li, B. Q.; Zhang, X. Z.; Wang, Z.; Zhang, K. Identification of bioactive compounds that contribute to the alpha-glucosidase inhibitory activity of rosemary. Food Funct. 2020, 11, 1692–1701. doi: 10.1039/C9FO02448D
Dan, W. J.; Zhang, Q.; Zhang, F.; Wang, W. W.; Gao, J. M. Benzonate derivatives of acetophenone as potent alpha-glucosidase inhibitors: synthesis, structure-activity relationship and mechanism. J. Enzyme Inhib. Med. Chem. 2019, 34, 937–945. doi: 10.1080/14756366.2019.1604519
Tong, T. T.; Zhao, E. H.; Gao, H. L.; Xu, Y. H.; Zhao, Y. J.; Fu, G.; Cui, H. J. Recent research advances of 1-deoxynojirimycin and its derivatives. China J. Chin. Mater. Med. 2018, 43, 1990–1997.
Paolini, J. P. The bond order-bond length relationship. J. Comput. Chem. 1990, 11, 1160–1163. doi: 10.1002/jcc.540111007
Cai, W.; Jiang, L.; Xie, Y.; Liu, Y.; Liu, W.; Zhao, G. Design of SGLT2 inhibitors for the treatment of type 2 diabetes: a history driven by biology to chemistry. Med. Chem. 2015, 11, 317–328. doi: 10.2174/1573406411666150105105529
Wang, Y.; Yao, H.; Hua, M.; Jiao, Y.; He, H.; Liu, M.; Huang, N.; Zou, K. Direct N-glycosylation of amides/amines with glycal donors. J. Org. Chem. 2020, 85, 7485–7493. doi: 10.1021/acs.joc.0c00975
Yao, Y.; Xiong, C. P.; Zhong, Y. L.; Bian, G. W.; Huang, N. Y.; Wang, L.; Zou, K. Intramolecular and Ferrier rearrangement strategy for the construction of C1-β-D-xylopyranosides: synthesis, mechanism and biological activity study. Adv. Syn. Cat. 2019, 361, 1012–1017. doi: 10.1002/adsc.201801423
Li, D. W.; Zuo, H. H.; Hu, M.; Zhang, J. Y.; Chen, L.; Huang, N. Y. Synthesis and absolute configuration of (2S, 3S, 3aS, 6S, 7aR)-2, 3-dihydroxy-2-((R)-1-hydroxy-3-methylbutyl)-3, 6-dimethylhexahydrobenzofuran-4(2H)-one. Chin. J. Struct. Chem. 2017, 36, 1276–1282.
Huang, N. Y.; Wang, W. B.; Chen, L.; Luo, H. J.; Wang, J. Z.; Deng, W. Q.; Zou, K. Design, synthesis and biological evaluation of bisabolonalone oxime derivatives as potassium-competitive acid blockers (P-CABs). Bioorg. Med. Chem. Lett. 2016, 26, 2268–2272. doi: 10.1016/j.bmcl.2016.03.051

Figure 1 ORTEP drawing of compound IX showing thermal ellipsoids at the 50% probability level

Figure 2 Packing diagram of compound IX. The O–H···O interactions are shown as dashed lines
Table 1. Optimization of Conditions for the C-glycosylation Reaction
| Entrya | Catalyst | P-Ligand | Solvent | Yieldb |
| 1 | Pd2(dba)3 | Xantphos | THF | 40% |
| 2 | Pd(OAc)2 | Xantphos | THF | 10% |
| 3 | White catalyst | Xantphos | THF | - |
| 4 | Pd(PPh3)4 | Xantphos | THF | - |
| 5 | PdCl2 | Xantphos | THF | - |
| 6 | Pd(acac)2 | Xantphos | THF | 60% |
| 7 | Pd(acac)2 | DPPB | THF | 86% |
| 8 | Pd(acac)2 | DPPF | THF | 74% |
| 9 | Pd(acac)2 | tBuXPhos | THF | 66% |
| 10 | Pd(acac)2 | P(Cy)3 | THF | 57% |
| 11 | Pd(acac)2 | DPPB | CH2Cl2 | 91% |
| 12 | Pd(acac)2 | DPPB | CH3CN | 88% |
| 13 | Pd(acac)2 | DPPB | Toluene | 76% |
| aUnless otherwise specified, all reactions were carried out with 0.1 mmol of IX, 0.2 mmol of nitromethane, 2.5 mol% Pd catalyst and 5 mol% P-ligand in 2 mL solvent and N2 atmosphere at room temperature. bIsolated yield, N.R. = No reaction. | ||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Selected Bond Lengths (Å) and Bond Angles (°) for Compound 3
| Bond | Dist. | Bond | Dist. | Bond | Dist. | ||
| C(1)–O(2) | 1.4383(17) | C(5)–C(6) | 1.518(2) | O(3)–C(7) | 1.4493(17) | ||
| O(2)–C(5) | 1.4233(17) | N(1)–C(6) | 1.493(2) | O(4)–C(8) | 1.1994(19) | ||
| C(5)–C(4) | 1.500(2) | O(6)–N(1) | 1.215(2) | O(5)–C(8) | 1.3280(19) | ||
| C(4)–C(3) | 1.325(2) | O(7)–N(1) | 1.2335(19) | O(1)–C(2) | 1.4305(19) | ||
| Angle | (°) | Angle | (°) | Angle | (°) | ||
| C(5)–O(2)–C(1) | 112.11(11) | O(6)–N(1)–O(7) | 124.16(15) | O(2)–C(1)–C(7)–O(3) | 83.57(13) | ||
| C(3)–C(4)–C(5) | 122.03(14) | O(4)–C(8)–O(3) | 125.22(15) | O(2)–C(1)–C(2)–O(1) | 74.93(15) | ||
| C(3)–C(4)–C(5) | 122.03(14) | C(8)–O(5)–C(9) | 121.05(12) | O(2)–C(1)–C(2)–C(3) | –49.34(16) | ||
| C(7)–C(1)–C(2) | 111.21(12) | C(11)–C(9)–C(10) | 112.40(15) | O(2)–C(5)–C(4)–C(3) | 11.5(2) |
下载: 导出CSV
Table 3. Hydrogen Bonds for Compound 3
| D–H⋅⋅⋅A | d(D–H) | d(H⋅⋅⋅A) | d(D⋅⋅⋅A) | < (DHA) |
| O(1)–H(1)⋅⋅⋅O(7)(a) | 0.82 | 2.08 | 2.8904(1) | 172 |
| C(4)–H(4)⋅⋅⋅O(4)(b) | 0.93 | 2.37 | 3.2683(1) | 163 |
| C(10)–H(10C)⋅⋅⋅O(4) | 0.96 | 2.37 | 2.9117(1) | 115 |
| Symmetry codes: (a) –1/2 + x, –1/2 – y, –z; (b) 1/2 – x, –y, –1/2 + z | ||||
下载: 导出CSV
Table 4. α-Glucosidase Inhibitory Activity of Compounds IV-IX
| Compound | IC50 (μg/mL) | Compound | IC50 (μg/mL) | |
| IV | > 200 | VIII | > 200 | |
| V | > 200 | IX | 89.86 | |
| VI | > 200 | 1-Deoxynojirimycina | 0.29 | |
| VII | > 200 | DMSOb | - | |
| aPositive control, bblank test. | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: