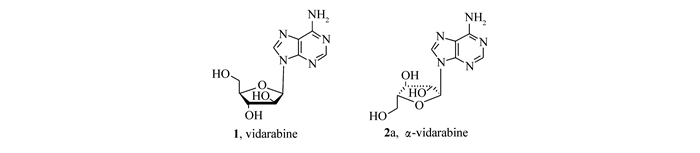
Scheme 1.
Structures of α-vidarabine(2a) and vidarabine(1)

核苷碱基的阿拉伯糖苷具有广泛的生理和药理活性,如阿拉伯糖腺苷(vidarabine,9-β-D-阿拉伯呋喃糖基腺嘌呤)广泛用于治疗疱疹病毒、巨细胞病毒、乙型肝炎病毒引起的疾病以及急性白血病[1]。从结构上来看,这些物质均为β构型,其差向异构体α-阿拉伯糖苷也是一类重要的活性物质。如α-阿拉伯糖腺苷(2a)是较好的抑制肿瘤细胞的先导药物(Scheme 1),具有和vidatabine类似的抗病毒活性[2]。α-构型核苷的结构存在于维生素B12及其同族维生素中[3]。其独特的结构特点也产生了独特的生物学性质[4]。但迄今为止,α-阿拉伯糖苷一直作为合成β构型的副产物,收率一般在5%以下[5]。α-阿拉伯糖苷的单一构型合成并未引起合成工作者的关注。
传统方法中糖苷键的生成均是在有机溶剂中,需要路易斯酸催化[6]。微波辅助的无溶剂、无催化剂条件具有操作简便、环境友好的优点,得到了有机化学工作者的密切关注,但是在糖苷键生成反应上的应用仅有一例非环核苷的报道[7]。
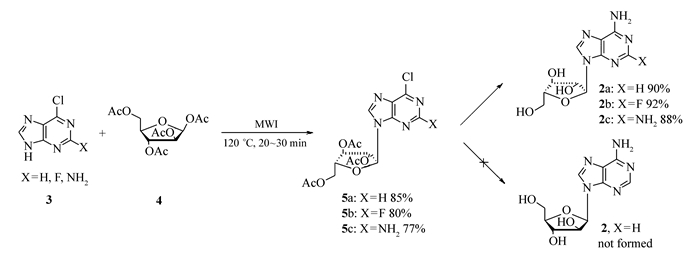
本文以廉价易得的卤代嘌呤(3)和1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖(4)为原料,在无溶剂、无催化剂条件下,加热熔融反应,即可生成糖苷键,得到缩合物,然后经脱除乙酰基、氨解等步骤,高选择性地得到α-腺嘌呤阿拉伯糖苷及其类似物,无β-异构体的产生(Scheme 2)。
6-氯嘌呤(≥98%)、2-氟-6-氯嘌呤(≥98%)、2-氨基-6-氯嘌呤(≥98%)、1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖(≥98%)购自新乡拓新医药股份有限公司;SnCl4、对甲苯磺酸(TsOH)、Triflic acid和其他常规溶剂购自上海阿拉丁生化科技股份有限公司,均为分析纯试剂;所用溶剂未经进一步处理。
AC 400型核磁共振仪(NMR, 德国Bruker公司),DMSO-d6或CDCl3为溶剂,四甲基硅烷(TMS)为内标;UPLC-QTOFMS型高分辨质谱(HRMS, 美国Waters公司);XRC-1型显微熔点仪(四川大学科仪厂,温度计未校正);MAS-Ⅰ型微波合成仪(上海新仪微波化学科技有限公司)。
6-氯嘌呤(0.308 g,2 mmol),1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖(0.763 g,2.4 mmol)加入到反应瓶中,混合均匀,置于微波反应器内,设定最高功率为200 W,温度120 ℃,反应20 min,降至室温,TLC检测原料基本消失,加入无水乙醇(50 mL),加热至溶解,加入活性炭(0.2 g)脱色,趁热过滤,滤液真空浓缩,得淡黄色油状物0.759 g,收率85%。
1H NMR(CDCl3, 400 MHz), δ:9.03(s, 1H), 8.41(s, 1H), 6.67(d, J=4.4 Hz, 1H), 5.49(t, J=4.0 Hz, 1H), 5.40(t, J=3.2 Hz, 1H), 4.44~4.36(m, 2H), 4.27~4.24(m, 1H), 2.09(s, 3H), 2.04(s, 3H), 1.79(s, 3H); 13C NMR(CDCl3, 100 MHz), δ:170.3, 169.6, 169.4, 152.8, 151.0, 149.0, 143.8, 134.6, 86.5, 80.4, 73.1, 70.5, 63.0, 20.8, 20.6, 20.4;HRMS计算值C16H17ClN4NaO7[M+Na]+ 435.0678, 实测值435.0678。
2-氟-6-氯嘌呤(0.346 g,2 mmol),1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖(0.763 g,2.4 mmol)加入到反应瓶中,混合均匀,置于微波反应器内,设定最高功率为200 W,温度120 ℃,反应25 min,降至室温,TLC检测原料基本消失,加入无水乙醇(50 mL),加热至溶解,加入活性炭(0.2 g)脱色,趁热过滤,滤液真空浓缩,得淡黄色油状物0.689 g,收率80%。
1H NMR(CDCl3, 400 MHz), δ:8.27(s, 1H), 6.48(d, J=4.4 Hz, 1H), 5.47~5.45(m, 1H), 5.34(t, J=3.6 Hz, 1H), 4.46~4.38(m, 2H), 4.27~4.23(m, 1H), 2.12(s, 3H), 2.08(s, 3H), 1.86(s, 3H); 13C NMR(CDCl3, 100 MHz), δ:170.3, 169.4, 168.4, 159.8(d, JC—F=215.6 Hz), 153.1(d, JC—F=16.2 Hz), 151.0(d, JC—F=15.5 Hz), 144.8(d, JC—F=2.8 Hz), 132.4(d, JC—F=4.2 Hz), 83.0, 80.2, 75.5, 74.6, 62.5, 20.6, 20.5, 20.1. HRMS计算值C16H16ClFN4NaO7[M+Na]+ 453.0584, 实测值453.0586。
2-氨基-6-氯嘌呤(0.338 g,2 mmol),1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖(0.763 g,2.4 mmol)加入到反应瓶中,混合均匀,置于微波反应器内,设定最高功率为200 W,温度120℃,反应30 min,降至室温,TLC检测原料基本消失,加入无水乙醇(50 mL),加热至溶解,加入活性炭(0.2 g)脱色,趁热过滤,滤液真空浓缩,得淡黄色油状物0.657 g,收率77%。
1H NMR(CDCl3, 400 MHz), δ:8.40(s, 1H), 6.66(d, J=4.4 Hz, 1H), 5.48(t, J=6.8 Hz, 1H), 5.39(t, J=3.2 Hz, 1H), 4.43~4.35(m, 2H), 4.26~4.23(m, 1H), 2.08(s, 3H), 2.03(s, 3H), 1.78(s, 3H); 13C NMR(CDCl3, 100 MHz), δ:170.3, 169.5, 169.3, 152.7, 150.9, 148.9, 143.7, 134.6, 86.4, 80.3, 73.0, 70.5, 62.9, 20.7, 20.5, 20.3;HRMS计算值C16H18ClN5NaO7[M+Na]+ 450.0787, 实测值450.0789。
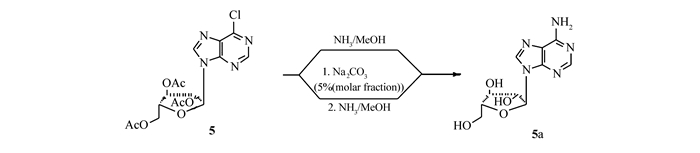
NH3/MeOH氨解法 上述化合物5a加入到高压反应罐中,加入饱和NH3/MeOH溶液(100 mL),密封,100 ℃反应10 h(反应罐压力:0.2 MPa),冷却至室温,缓慢释放压力,反应液减压浓缩,得到淡黄色油状物,加入无水乙醇(20 mL),加热溶解,加入活性炭(0.1 g)脱色,趁热过滤,滤液减压浓缩,得到白色固体2a 0.95 g,收率79%。
一锅法 上述化合物5a加入到高压反应罐中,用MeOH(50 mL)溶解,加入Na2CO3(5.3 mg),室温搅拌1 h,TLC检测原料5a消失,再加入饱和NH3/MeOH溶液(50 mL),密封,100 ℃反应10 h(反应罐压力:0.1 MPa),冷却至室温,缓慢释放压力,反应液减压浓缩,得到淡黄色固体,加入无水乙醇(20 mL),加热溶解,加入活性炭(0.1 g)脱色,趁热过滤,滤液减压浓缩,得到白色固体2a 1.2 g,收率92%。纯度>98%[HPLC归一化法:色谱柱Kromasil C18柱(4.6 mm×150 mm,5 μm);流动相(V(水):V(乙腈)=1:1);检测波长254 nm;流速1 mL/min;进样量10 μL]。
白色固体,mp 212~214 ℃(文献[8] mp 213 ℃)。1H NMR(DMSO-d6, 400 MHz), δ:8.35(s, 1H), 8.14(s, 1H), 7.37(brs, 2H), 6.02(d, J=4.4 Hz, 1H), 5.48(t, J=6.0 Hz, 2H), 5.23(d, J=4.4 Hz, 1H), 4.64~4.59(m, 1H), 4.16~4.13(m, 1H), 3.98~3.96(m, 1H), 3.70~3.53(m, 2H); 13C NMR(DMSO-d6, 100 MHz), δ:155.9, 152.4, 149.4, 140.3, 118.2, 84.0, 83.6, 75.6, 74.9, 60.8;HRMS计算值C10H14N5O4[M+H]+ 268.1040, 实测值268.1046。
类似方法合成化合物2b和2c。
α-2-氟腺嘌呤阿拉伯糖苷(2b) 白色固体,mp 223~225 ℃。1H NMR(DMSO-d6, 400 MHz), δ:8.17(s, 1H), 7.75(brs, 2H), 6.06(d, J=4.4 Hz, 1H), 5.66(d, J=5.6 Hz, 1H), 5.57(d, J=4.8 Hz, 1H), 5.12(d, J=5.2 Hz, 1H), 4.18~4.09(m, 2H), 3.79~3.76(m, 1H), 3.68~3.49(m, 2H); 13C NMR(DMSO-d6, 100 MHz), δ:159.6(d, JC—F =230 Hz), 157.6(d, JC—F=3.1 Hz), 150.8(d, JC—F=20 Hz), 140.6, 116.6(d, JC—F=4 Hz), 84.1, 83.8, 75.6, 74.8, 60.8;HRMS计算值C10H13FN5O4[M+H]+ 286.0946, 实测值286.0948。
α-2-氨基腺嘌呤阿拉伯糖苷(2c) 白色固体,mp 246~248 ℃。1H NMR(DMSO-d6, 400 MHz), δ:7.79(s, 1H), 6.66(brs, 2H), 6.13(d, J=4.4 Hz, 1H), 5.76(brs, 2H), 5.64(d, J=5.6 Hz, 1H), 5.52(d, J=4.4 Hz, 1H), 5.13(t, J=5.2 Hz, 1H), 4.09~4.03(m, 2H), 3.74(t, J=4.4 Hz, 1H), 3.66~3.46(m, 2H); 13C NMR(DMSO-d6, 100 MHz), δ:160.2, 156.0, 151.6, 137.1, 112.4, 84.1, 83.3, 75.6, 61.1;HRMS计算值C10H15N6O4[M+H]+ 283.1149, 实测值283.1153。
以6-氯嘌呤和1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖的缩合为模板反应,考查溶剂、催化剂、投料比、反应时间、反应温度及反应规模对缩合产物收率的影响。结果见表 1。
 下载:
导出CSV
下载:
导出CSV
| Entry | n(3): n(4) | Solvent | Catalyst(equiv.) | Temperture/℃ | Reaction time/min | Yield/%c |
| 1 | 1:1 | Cl(CH2)2Cl | SnCl4(0.2) | 80 | 120 | 32 |
| 2 | 1:1 | Cl(CH2)2Cl | TsOH(0.2) | 80 | 120 | 31 |
| 3 | 1:1 | Cl(CH2)2Cl | Triflic acid(0.2) | 80 | 120 | 42 |
| 4 | 1:1 | CH3CN | Triflic acid(0.2) | 80 | 120 | 29 |
| 5 | 1:1 | C6H5Cl | Triflic acid(0.2) | 80 | 120 | 19 |
| 6 | 1:1 | 0 | Triflic acid(0.2) | 80 | 120 | 57 |
| 7 | 1:1 | 0 | 0 | 80 | 120 | 62 |
| 8 | 1:1 | 0 | 0 | 100 | 120 | 65 |
| 9 | 1:1 | 0 | 0 | 120 | 120 | 70 |
| 10 | 1:1.2 | 0 | 0 | 120 | 120 | 75 |
| 11 | 1:1.5 | 0 | 0 | 120 | 120 | 76 |
| 12 | 1:1.2 | 0 | 0 | 120 | 120 | 78 |
| 13 | 1:1.2 | 0 | 0 | 120 | 120 | 80 |
| 14d | 1:1.2 | 0 | 0 | 120 | 20 | 85 |
| 15d | 1:1.2 | 0 | 0 | 120 | 25 | 83 |
| a.3(0.308 g, 2 mmol), solvent(5 mL); b.entries 1~14, 120 min, entry 15, 20 min, entry 16, 25 min; c.isolated yield; d.MWI, 0~200 W. | ||||||
由表 1可知,n(3):n(4)=1:1,以二氯乙烷为溶剂,80 ℃反应2 h,以0.2倍化学计量的SnCl4为催化剂,收率为32%。催化剂改变为TsOH时,收率相当。当催化剂改变为Triflic acid时,收率提高到42%。再进一步筛选溶剂,发现乙腈和氯苯均不能很好反应。令人高兴的是,当不使用溶剂,直接将原料混合熔融反应,收率可以提高到57%。如果不加入催化剂,收率稍有增加,达62%。于是在无催化剂和无溶剂条件下继续优化反应条件。反应温度升高可以促进反应的发生。当反应温度为120 ℃时,收率达70%。继续升高温度,会使原料变黑碳化,同时会造成操作不便,未进一步升高温度。进一步地,增加原料4的比例至1.2倍化学计量,收率提高到75%,继续增加至1.5倍化学计量,收率基本不再增加,所以以n(3):n(4)=1:1.2为最优。进一步,尝试在微波辐射下进行该反应,20 min时收率即可以得到85%,显示出微波对反应有较好的促进作用,继续延长反应时间,收率不再增加,所以以微波辐射下120 ℃的反应为最优。综上,最优的反应条件为:n(3):n(4)=1:1.2,微波辐射,120 ℃,20 min,收率85%。
参考文献[9],缩合物5a在饱和NH3/MeOH溶液中高压反应,在脱除糖环上的乙酰基的同时,使6-Cl氨解。但是在反应的同时产生3倍化学计量的乙酰胺,乙酰胺溶解在溶剂中,使产物难以析出。为了解决这个问题,先加入相对于缩合物5a摩尔分数为5%的Na2CO3固体,室温搅拌1 h,酯交换脱除乙酰基,然后不需要分离,直接加入饱和NH3/MeOH溶液,高压氨解,得到产物2a,这样避免了产生过量的乙酰胺,产物2a很容易从反应体系中析出,不需要柱层析(Scheme 3)。
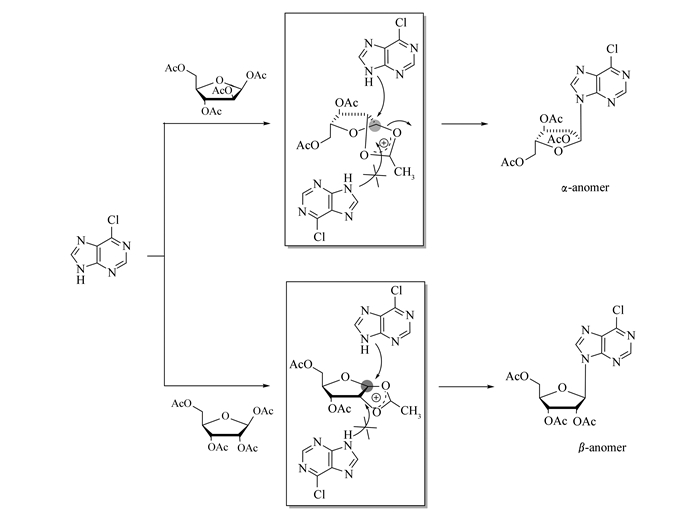
得到的产品均是α-构型,糖基1′位氢与2′位氢偶合常数J≈4.0 Hz。因为1, 2, 3, 5-四-O-乙酰基-β-D-核糖与6-氯嘌呤缩合,得到的是单一的β构型产物,而1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖与6-氯嘌呤缩合,得到的是单一的α构型产物。因此,可能的反应机理是:1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖在加热后,生成环鎓离子,由于2位羟基的位阻效应,6-氯嘌呤单一性地从α位进攻端头碳,得到α-构型产物(Scheme 4)。原料3中吸电子基团Cl或F原子对于缩合反应非常关键,带有给电子基团氨基或甲氧基等,反应不能发生。
发展了一种在无溶剂、无催化剂条件下有效合成α-腺嘌呤阿拉伯糖苷及其类似物的方法。以卤代嘌呤和1, 2, 3, 5-四-O-乙酰基-β-D-阿拉伯糖为原料,在微波辐射下,无溶剂、无催化剂地在熔融状态发生缩合反应,得到缩合物;缩合物在Na2CO3催化下脱除乙酰基,“一锅”加入饱和NH3/CH3OH溶液氨解,以2步和76%的总收率得到α-腺嘌呤阿拉伯糖苷。缩合反应可以扩大到100 g的反应规模,收率没有降低。2-氟-6-氯嘌呤、2-氨基-6-氯嘌呤也能很好地反应。推测的反应机理显示,碱基从位阻较小的α位进攻环鎓离子,从而单一性地得到α构型产物。对比传统方法,该方法避免使用重金属催化剂和有机溶剂,操作简便,中间体及产物可以通过结晶的方法纯化得到,为α-腺嘌呤阿拉伯糖苷及其类似物的合成及活性研究提供了有益的参考,同时显示出潜在的应用价值。
辅助材料(Supporting Information)[产物及中间体的1H NMR和13C NMR图谱]可以免费从本刊网站(http://yyhx.ciac.jl.cn/)下载。
Theobald R J. Vidarabine[M]. 2016, Elsevier Inc.
Revankar G R, Huffman J H, Allen L B. Synthesis and Antiviral Activity of Certain 5'-Monophosphates of 9-D-Arabinofuranosyladenine and 9-D-Arabinofuranosylhypoxanthine[J]. J Med Chem, 1975, 18(7): 721-726. doi: 10.1021/jm00241a016
Thomas H J, Montgomery J A. 7-Glycosylpurines.Ⅱ.Arabinofuranosides of Hypoxanthine and Adenine[J]. J Org Chem, 1966, 31(5): 1413-1416. doi: 10.1021/jo01343a023
York J L. Effect of the Structure of the Glycon on the Acid-catalyzed Hydrolysis of Adenine Nucleosides[J]. J Org Chem, 1981, 46(10): 2171-2173. doi: 10.1021/jo00323a040
Lee W W, Benitez A, Goodman L. Potential Anticancer Agent:Synthesis of the β-Anomer of 9-(D-Arabinofuranosyl) Adenine[J]. J Am Chem Soc, 1960, 82(10): 2648-2649. doi: 10.1021/ja01495a070
Bookser B C, Raffaele N B. High-Throughput Five Minute Microwave Accelerated Glycosylation Approach to the Synthesis of Nucleoside Libraries[J]. J Org Chem, 2007, 72(1): 173-179.
Qu G R, Han S H, Zhang Z G. Microwave-assisted Regioselective Synthesis of Acyclic Nucleosides Through an Alkylating Reaction with 2-Axa-1, 4-butanediol Diacetate[J]. Can J Chem, 2006, 84(5): 819-824. doi: 10.1139/v06-061
Montgomery J A, Laseter A G, Shortnacy A T. Nucleosides of 2-Azapurines. 7H-Imidazo[4, 5-d]-1, 2, 3-triazines[J]. J Med Chem, 1975, 18(6): 564-567. doi: 10.1021/jm00240a006
Janeba Z, Francom P, Robins M J. Efficient Syntheses of 2-Chloro-2-deoxyadenosine(Cladribine) from 2-Deoxyguanosine[J]. J Org Chem, 2003, 68(3): 989-992. doi: 10.1021/jo020644k
表 1 反应条件优化a
Table 1. Optimization of reaction conditionsa
| Entry | n(3): n(4) | Solvent | Catalyst(equiv.) | Temperture/℃ | Reaction time/min | Yield/%c |
| 1 | 1:1 | Cl(CH2)2Cl | SnCl4(0.2) | 80 | 120 | 32 |
| 2 | 1:1 | Cl(CH2)2Cl | TsOH(0.2) | 80 | 120 | 31 |
| 3 | 1:1 | Cl(CH2)2Cl | Triflic acid(0.2) | 80 | 120 | 42 |
| 4 | 1:1 | CH3CN | Triflic acid(0.2) | 80 | 120 | 29 |
| 5 | 1:1 | C6H5Cl | Triflic acid(0.2) | 80 | 120 | 19 |
| 6 | 1:1 | 0 | Triflic acid(0.2) | 80 | 120 | 57 |
| 7 | 1:1 | 0 | 0 | 80 | 120 | 62 |
| 8 | 1:1 | 0 | 0 | 100 | 120 | 65 |
| 9 | 1:1 | 0 | 0 | 120 | 120 | 70 |
| 10 | 1:1.2 | 0 | 0 | 120 | 120 | 75 |
| 11 | 1:1.5 | 0 | 0 | 120 | 120 | 76 |
| 12 | 1:1.2 | 0 | 0 | 120 | 120 | 78 |
| 13 | 1:1.2 | 0 | 0 | 120 | 120 | 80 |
| 14d | 1:1.2 | 0 | 0 | 120 | 20 | 85 |
| 15d | 1:1.2 | 0 | 0 | 120 | 25 | 83 |
| a.3(0.308 g, 2 mmol), solvent(5 mL); b.entries 1~14, 120 min, entry 15, 20 min, entry 16, 25 min; c.isolated yield; d.MWI, 0~200 W. | ||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: