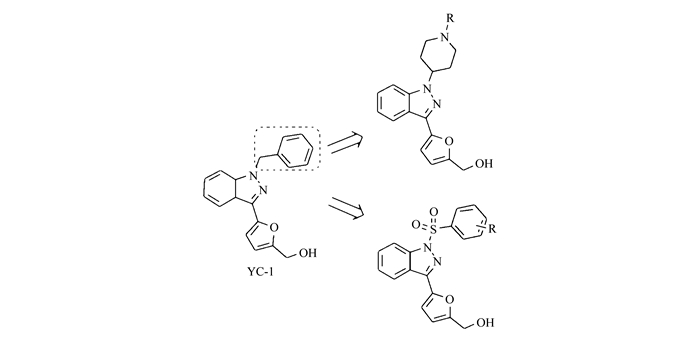
图 1
先导化合物及设计的目标化合物结构
Figure 1.
The structures of lead compounds and designed compounds

肝细胞癌(Hepatocellular carcinoma, HCC)是全球最常见的肿瘤之一,越来越多的研究发现,炎性微环境在包括肝癌在内的多种肿瘤进展、血管生成和侵袭中发挥了重要作用[1]。低氧诱导因子l(HIF-1)是调控肿瘤内缺氧效应的关键转录因子,包括HIF-1α和HIF-1β。它能控制肿瘤细胞在缺氧条件下众多基因的表达,改变肿瘤细胞内糖代谢的方式,增强肿瘤组织血管生成,促进肿瘤细胞的增殖、侵袭及转移。由于细胞内对HIF-1的调控主要通过其α亚基进行,因此HIF-1α抑制剂成为抗肿瘤药物的研究热点。HIF-1α能够在3个方面加快糖酵解:1)HIF-1α转录表达出的与线粒体相关的酶类(如丙酮酸脱氢酶激酶1, PDK1等);2)HIF-1α的表达上调其下游的葡萄糖转运蛋白靶基因(GLUT1和GLUT3);3)激活转录与葡萄糖代谢和糖酵解相关的关键酶类(如磷酸甘油酸酯激酶1, PGK1等)[2]。其中,HIF抑制剂3-(5′-羟甲基-2′-呋喃基)-1-苯甲基吲唑(YC-1)因抑制HIF-1α表达及影响HIF-1α稳定性,而表现出潜在的抗肿瘤特性[3-4]。
近年来,有大量的磺酰胺类抗肿瘤药物的研究发表。Lobb等[5]将已有的抑制血管生成的磺酰胺类药物LY32262和LY33169结构进行改造获得了抗肿瘤活性更强的新化合物。刘菲等[6]设计合成的一系列磺酰脒化合物也具有良好的抗肺癌、肝癌及结肠癌的作用。此外,在本课题组的前期研究中发现,在改造结构中连接哌啶基不仅能够提高化合物的稳定性,还能够调节化合物的pH值[7-8]。
本文在前期研究工作的基础上,以HIF抑制剂YC-1为先导化合物,经优化药效基团设计合成了9个包含磺酰和哌啶结构的1, 3-取代吲唑类化合物。所合成的化合物采用四甲基偶氮唑蓝(MTT)法初步评价了其对肝癌HepG2细胞增殖的体外抑制活性。其先导化合物及设计的目标化合物结构如图 1所示。
AVⅢ型600 MHz核磁共振谱仪(瑞士Bruker公司); Q-Exactive型质谱仪(美国Thermo Fisher公司);AB3200Q型TRAP-MS质谱仪(美国AB公司);Victor3 V型多功能酶标仪(美国Perkin-Elmer公司);CEM DISCOVER环形聚焦单模微波合成系统(美国CEM公司)。
细胞株HepG2(中国科学院典型培养物保藏委员会细胞库),实验室自行传代培养。1-Boc-4-甲烷磺酰氧基哌啶(≥98%,北京伊诺凯科技有限公司)、N, N-二甲基甲酰胺(≥98%,DMF, 上海泰坦科技股份有限公司)、硅胶(青岛海洋化工集团),其它所用试剂均为市售化学纯或分析纯。
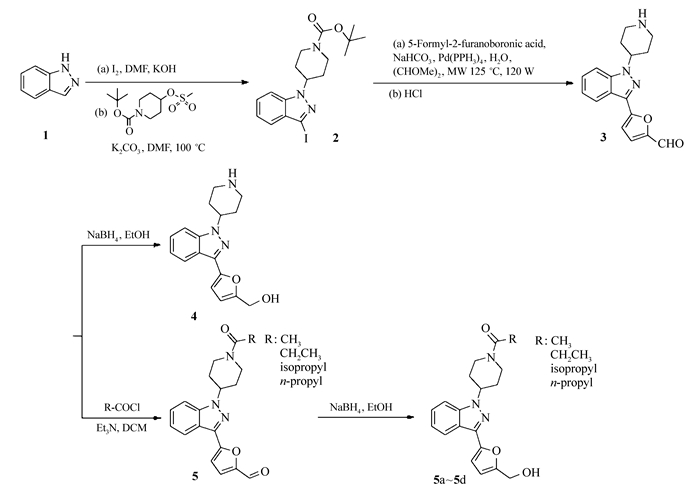
将1.00 g(8.46 mmol)吲唑与2.37 g(9.32 mmol)碘晶体溶解于10 mL DMF中,加入饱和氢氧化钠溶液5 mL,室温搅拌3 h。反应完毕后,加水、乙酸乙酯萃取3次,有机层用无水硫酸钠干燥,减压蒸除溶剂。除去溶剂后,将2.05 g(8.40 mmol)中间产物与2.37 g(8.48 mmol) 1-Boc-4-甲烷磺酰氧基哌啶再次溶于10 mL DMF中,在冰浴下加入碳酸钾搅拌10 min后,在油浴中加热到100 ℃,搅拌过夜。反应后加水、乙酸乙酯萃取3次,有机层依次用稀盐酸溶液、纯水、饱和氯化钠溶液分别萃取2次,后用无水硫酸钠干燥,减压蒸除溶剂,经硅胶柱层析(V(乙酸乙酯):V(正己烷)=1:10)分离得获得淡黄色固体(中间体2)。将3.50 g(8.20 mmol)中间体2,1.38 g(9.86 mmol) 5-甲醛基呋喃-2-硼酸和0.95 g(0.82 mmol)四(三苯基膦)钯溶解于50 mL乙二醇二甲醚(DME)中,加入5 mL饱和碳酸氢钠溶液,在微波反应器中以125 ℃,120 W的条件反应15 min。用水、乙酸乙酯萃取3次,有机层用饱和氯化钠溶液萃取2次,减压蒸除溶剂。将其溶于20 mL乙酸乙酯,缓慢滴入5~6滴浓盐酸,室温搅拌4 h,除去溶剂后,经硅胶柱层析(V(甲醇):V(二氯甲烷)=1:10)分离得获得1.87 g白色固体(中间体3),收率74.8%。
将2.36 g(8.00 mmol)中间体3与1.21 g(3.20 mmol)硼氢化钠在干燥的Ar气条件下,缓慢滴入15 mL无水乙醇,室温搅拌10 h。反应完成后,加入稀盐酸溶液调节反应液pH值至7,抽滤去除沉淀,减压蒸干溶剂,经硅胶柱层析(V(甲醇):V(二氯甲烷)=1:8)获得1.40 g白色固体(化合物4),收率56%。1H NMR(600 MHz, DMSO-d6), δ:8.20(d, J=8.25 Hz, 1H), 7.93(d, J=8.25 Hz, 1H), 7.75(d, J=3.85 Hz, 1H), 7.56(dt, J=0.73, 7.70 Hz, 1H), 7.38(t, J=7.61 Hz, 1H), 7.33(d, J=3.85 Hz, 1H), 5.14~5.21(m, 1H), 3.43~3.49(m, 3H), 3.14~3.21(m, 3H), 2.39~2.48(m, 3H), 2.12~2.22(m, 3H)。MS(ESI)计算值C17H19N3O2[M+H]+:298.1, 实测值:298.1。
将1.00 g(3.39 mmol) 3-(5′-甲醛-2′-呋喃)-1-哌啶基吲唑(中间体3)溶于5 mL二氯甲烷(DCM), 加入0.40 g(5.09 mmol)乙酰氯,缓慢滴入0.5 mL三乙胺,室温搅拌3 h。反应后,加水、二氯甲烷萃取2次,有机层用饱和食盐水萃取2次后,减压旋蒸。经硅胶层析柱(V(乙酸乙酯):V(正己烷)=1:2)分离得到1.10 g白色固体。将1.10 g(3.26 mmol)白色固体在干燥的环境下,溶于无水乙醇,在冰浴下搅拌5 min,缓慢加入0.37 g(9.78 mmol)硼氢化钠,持续冰浴搅拌10 min后恢复室温,搅拌3 h。充分反应后,加入稀盐酸溶液淬灭,调pH值至7,抽滤除去沉淀,减压旋蒸。经硅胶层析柱(V(甲醇):V(DCM)=1:10)分离得到0.74 g白色固体,收率为64%。mp 243.3~244.6 ℃。1H NMR(600 MHz, CDCl3), δ:8.09(d, J=8.25 Hz, 1H), 7.38~7.48(m, 3H), 7.23(ddd, J=1.19, 6.56, 8.02 Hz, 1H), 6.83(d, J=3.30 Hz, 1H), 6.46(d, J=3.30 Hz, 1H), 4.78(d, J=13.76 Hz, 1H), 4.74(s, 2H), 4.65 ~ 4.69(m, 1H), 4.03(d, J=14.12 Hz, 1H), 3.25~3.34(m, 1H), 2.80~2.88(m, 1H), 2.37(dq, J=4.31, 12.20 Hz, 1H), 2.26(dq, J=4.31, 12.20 Hz, 1H), 2.16(s, 3H), 2.07~2.13(m, 2H); 13C NMR(151 MHz, CDCl3), δ:169.0, 153.9, 148.8, 139.6, 136.1, 126.6, 121.9, 121.4, 121.3, 109.6, 108.9, 107.7, 57.6, 56.1, 45.6, 40.8, 31.7, 31.1, 21.4。MS(ESI)计算值C19H21N3O3[M+H]+:340.2, 实测值:340.2。
将1.00 g(3.39 mmol) 3-(5″-甲醛-2′-呋喃)-1-哌啶基吲唑(中间体3)溶于5 mL DCM, 加入0.47 g(5.09 mmol)丙酰氯,缓慢滴入0.5mL三乙胺,室温搅拌3 h。反应后,加水、二氯甲烷萃取2次,有机层用饱和食盐水萃取2次后,减压旋蒸。经硅胶层析柱(V(乙酸乙酯):V(正己烷)=1:2)分离得到1.08 g白色固体。将1.08 g(3.09 mmol)白色固体在干燥的环境下,溶于无水乙醇,在冰浴下搅拌5 min,缓慢加入0.35 g(9.27 mmol)硼氢化钠, 持续冰浴搅拌10 min后恢复室温,搅拌3 h。充分反应后,加入稀盐酸溶液淬灭,调pH值至7,抽滤除去沉淀,减压旋蒸。经硅胶层析柱(V(甲醇):V(DCM)=1:10)分离得到0.73 g白色固体,收率为61%。mp 249.8~250.6 ℃。1H NMR(600 MHz, CDCl3), δ:8.08(d, J=8.25 Hz, 1H), 7.39~7.45(m, 2H), 7.22(t, J=7.24 Hz, 1H), 6.83(d, J=3.30 Hz, 1H), 6.45(d, J=3.30 Hz, 1H), 4.80(d, J=13.02 Hz, 1H), 4.74(s, 2H), 4.66(tt, J=4.17, 11.14 Hz, 1H), 4.07(d, J=13.75 Hz, 1H), 3.25(t, J=12.75 Hz, 1H), 2.83(t, J=12.20 Hz, 1H), 2.41(q, J=7.34 Hz, 2H), 2.27~2.38(m, 2H), 2.16(s, 1H), 2.04~2.13(m, 2H), 1.19(t, J=7.43 Hz, 3H); 13C NMR(151 MHz, CDCl3), δ:172.3, 154.0, 148.9, 139.7, 136.1, 126.6, 121.9, 121.4, 121.3, 109.6, 109.0, 107.7, 57.7, 56.3, 44.7, 41.0, 31.8, 31.2, 26.6, 9.6。MS(ESI)计算值C20H23N3O3[M+H]+:354.2, 实测值:354.2。
将1.00 g(3.39 mmol) 3-(5′-甲醛-2′-呋喃)-1-哌啶基吲唑(中间体3)溶于5 mL DCM, 加入0.54 g(5.09 mmol)异丁酰氯,缓慢滴入0.5 mL三乙胺,室温搅拌3 h。反应后,加水、二氯甲烷萃取2次,有机层用饱和食盐水萃取2次后,减压旋蒸。经硅胶层析柱(V(乙酸乙酯):V(正己烷)=1:2)分离得到1.07 g白色固体。将1.07 g(2.96 mmol)白色固体在干燥的环境下,溶于无水乙醇,在冰浴下搅拌5 min,缓慢加入0.34 g(8.88 mmol)硼氢化钠, 持续冰浴搅拌10 min后恢复室温,搅拌3 h。充分反应后,加入稀盐酸溶液淬灭,调pH值至7,抽滤除去沉淀,减压旋蒸。经硅胶层析柱(V(甲醇):V(DCM)=1:10)分离得到0.74 g白色固体,收率为59%。mp 215.0~216.0 ℃。1H NMR(600 MHz, CDCl3), δ:8.08(d, J=8.25 Hz, 1H), 7.39~7.45(m, 2H), 7.23(dt, J=0.83, 7.29 Hz, 1H), 6.83(d, J=3.30 Hz, 1H), 6.46(d, J=3.12 Hz, 1H), 4.82(d, J=12.65 Hz, 1H), 4.74(d, J=5.50 Hz, 2H), 4.68(tt, J=4.24, 11.26 Hz, 1H), 4.16(d, J=12.84 Hz, 1H), 3.28(t, J=12.20 Hz, 1H), 2.79~2.90(m, 2H), 2.36(d, J=10.64 Hz, 1H), 2.26(d, J=10.45 Hz, 1H), 2.16~2.20(m, 1H), 2.06~2.16(m, 2H), 1.14~1.22(m, 6H); 13C NMR(151 MHz, CDCl3), δ:175.5, 153.9, 148.9, 139.7, 136.1, 126.6, 121.9, 121.4, 121.3, 109.6, 109.0, 107.7, 57.7, 56.4, 44.6, 41.1, 32.0, 31.3, 30.2, 19.6, 19.4。MS(ESI)计算值C21H25N3O3[M+H]+:368.2, 实测值:368.2。
将1.00 g(3.39 mmol) 3-(5′-甲醛-2′-呋喃)-1-哌啶基吲唑(中间体3)溶于5 mL DCM, 加入0.54 g(5.09 mmol)丁酰氯,缓慢滴入0.5 mL三乙胺,室温搅拌3 h。反应后,加水、二氯甲烷萃取2次,有机层用饱和食盐水萃取2次后,减压旋蒸。经硅胶层析柱(V(乙酸乙酯):V(正己烷)=1:2)分离得到1.05 g白色固体。将1.05 g(2.87 mmol)白色固体在干燥的环境下,溶于无水乙醇,在冰浴下搅拌5 min,缓慢加入0.33 g(8.61 mmol)硼氢化钠, 持续冰浴搅拌10 min后恢复室温,搅拌3 h。充分反应后,加入稀盐酸溶液淬灭,调pH值至7,抽滤除去沉淀,减压旋蒸。经硅胶层析柱(V(甲醇):V(DCM)=1:10)分离得到0.73 g白色固体,收率为59%。mp 205.9~207.1 ℃。1H NMR(600 MHz, CDCl3), δ:8.08(d, J=8.25 Hz, 1H), 7.43~7.45(m, 1H), 7.39~7.42(m, 1H), 7.23(ddd, J=1.19, 6.56, 8.02 Hz, 1H), 6.83(d, J=3.30 Hz, 1H), 6.46(d, J=3.30 Hz, 1H), 4.80(d, J=13.57 Hz, 1H), 4.74(s, 2H), 4.67(tt, J=4.17, 11.23 Hz, 1H), 4.05~4.11(m, 1H), 3.22~3.29(m, 1H), 2.79~2.86(m, 1H), 2.35~2.40(m, 3H), 2.20~2.35(m, 2H), 2.07~2.14(m, 2H), 1.71(m, 2H), 1.00(t, J=7.43 Hz, 3H); 13C NMR(151 MHz, CDCl3), δ:171.6, 153.9, 148.9, 139.7, 136.1, 126.6, 121.9, 121.4, 121.3, 109.6, 109.0, 107.7, 57.7, 56.3, 44.9, 40.9, 35.4, 31.8, 31.2, 18.8, 14.1。MS(ESI)计算值C21H25N3O3[M+H]+:368.2, 实测值:368.2。
将1.00 g(8.46 mmol)吲唑与2.37 g(9.32 mmol)碘晶体溶解于10 mL DMF中,加入饱和氢氧化钠溶液5 mL,室温搅拌3 h。反应完毕后,加水、乙酸乙酯萃取3次,有机层用无水硫酸钠干燥,减压蒸除溶剂。除去溶剂后,将1.98 g(8.11 mmol)中间体,1.25 g(8.92 mmol) 5-甲醛基呋喃-2-硼酸和0.94 g(0.81 mmol)四(三苯基膦)钯溶解于50 mL DME中,加入5 mL饱和碳酸氢钠溶液,在微波反应器中以125 ℃,120 W的条件反应15 min。加水、乙酸乙酯萃取3次,有机层用饱和氯化钠溶液萃取2次,减压蒸除溶剂。经硅胶柱层析(V(乙酸乙酯):V(正己烷)=1:5)分离得获得1.36g白色固体(中间体6),收率76%。1H NMR(600 MHz, DMSO-d6), δ:13.72(s, 1H), 9.68(s, 1H), 8.17(d, J=8.25 Hz, 1H), 7.73(d, J=3.67 Hz, 1H), 7.66(d, J=8.44 Hz, 1H), 7.48(ddd, J=0.92, 7.01, 8.21 Hz, 1H), 7.32(ddd, J=0.73, 7.02, 8.02 Hz, 1H), 7.29(d, J=3.67 Hz, 1H); 13C NMR(151 MHz, DMSO-d6), δ:178.2, 154.6, 151.9, 141.5, 135.0, 127.4, 125.6, 122.7, 120.9, 120.3, 111.4, 109.5。
将1.00 g(4.72 mmol) 3-(5′-甲醛-2′-呋喃)-吲唑(中间体6)溶于5 mL DCM, 加入1.25 g(7.08 mmol)苯磺酰,缓慢滴入1 mL三乙胺,室温搅拌6 h。反应后,加水、二氯甲烷萃取2次,有机层用饱和食盐水萃取2次后,减压旋蒸。经硅胶层析柱(V(甲醇):V(二氯甲烷)=1:2)分离得到1.61 g白色固体。将1.61 g(4.57 mmol)白色固体在干燥的环境下,溶于无水乙醇,在冰浴下搅拌5 min,缓慢加入0.52 g(13.71 mmol)硼氢化钠, 持续冰浴搅拌10 min后恢复室温,搅拌3 h。充分反应后,加入稀盐酸溶液淬灭,调pH值至7,抽滤除去沉淀,减压旋蒸。经硅胶层析柱(V(乙酸乙酯):V(正己烷)=1:2)分离得到1.05 g白色固体,收率为63%。mp 198.6~199.5 ℃。1H NMR(600 MHz, CDCl3), δ:8.22(d, J=8.62 Hz, 1H), 8.09(d, J=8.07 Hz, 1H), 7.98~8.00(m, 2H), 7.58(ddd, J=0.92, 7.24, 8.34 Hz, 1H), 7.51 ~ 7.55(m, 1H), 7.41 ~ 7.45(m, 2H), 7.36~7.40(m, 1H), 7.07(d, J=3.30 Hz, 1H), 6.47(d, J=3.48 Hz, 1H), 4.73(s, 2H), 1.70(br s, 1H); 13C NMR(151 MHz, CDCl3), δ:155.7, 146.8, 143.3, 141.4, 137.4, 134.1, 129.5, 129.2, 127.5, 124.7, 123.4, 122.3, 113.4, 111.5, 109.8, 57.6。
将1.00 g(4.72 mmol) 3-(5′-甲醛-2′-呋喃)-吲唑(中间体6)溶于5 mL DCM, 加入1.35 g(7.08 mmol)对甲苯磺酰,缓慢滴入1 mL三乙胺,室温搅拌6 h。反应后,加水、二氯甲烷萃取2次,有机层用饱和食盐水萃取2次后,减压旋蒸。经硅胶层析柱(V(甲醇):V(DCM)=1:2)分离得到1.52 g白色固体。将1.52 g(4.15 mmol)白色固体在干燥的环境下,溶于无水乙醇,在冰浴下搅拌5 min,缓慢加入0.47 g(12.45 mmol)硼氢化钠, 持续冰浴搅拌10 min后恢复室温,搅拌3 h。充分反应后,加入稀盐酸溶液淬灭,调pH值至7,抽滤除去沉淀,减压旋蒸。经硅胶层析柱(V(乙酸乙酯):V(正己烷)=1:2)分离得到1.01 g白色固体,收率为58%。mp 203.5~204.5 ℃。1H NMR(600 MHz, CDCl3), δ:8.21(d, J=8.44 Hz, 1H), 8.08(d, J=8.07 Hz, 1H), 7.86(d, J=8.25 Hz, 2H), 7.51~7.60(m, 1H), 7.37(t, J=7.52 Hz, 1H), 7.21(d, J=8.25 Hz, 2H), 7.06(d, J=3.48 Hz, 1H), 6.46(d, J=3.30 Hz, 1H), 4.73(s, 2H), 2.32(s, 3H), 1.73(br s, 1H); 13C NMR(151 MHz, CDCl3), δ:155.6, 146.8, 145.4, 143.2, 141.4, 134.5, 129.8, 129.4, 127.6, 124.6, 123.4, 122.2, 113.4, 111.3, 109.8, 57.6, 21.6。HRMS(ESI)计算值C19H16N2O4S [M+H]+:369.0911, 实测值:369.0909。
将1.00 g(4.72 mmol) 3-(5′-甲醛-2′-呋喃)-吲唑(中间体6)溶于5 mL DCM, 加入1.35 g(7.08 mmol)间甲苯磺酰,缓慢滴入1 mL三乙胺,室温搅拌6 h。反应后,加水、二氯甲烷萃取2次,有机层用饱和食盐水萃取2次后,减压旋蒸。经硅胶层析柱(V(甲醇):V(DCM)=1:2)分离得到1.37 g白色固体。将1.37 g(3.74 mmol)白色固体在干燥的环境下,溶于无水乙醇,在冰浴下搅拌5 min,缓慢加入0.42 g(11.22 mmol)硼氢化钠, 持续冰浴搅拌10 min后恢复室温,搅拌3 h。充分反应后,加入稀盐酸溶液淬灭,调pH值至7,抽滤除去沉淀,减压旋蒸。经硅胶层析柱(V(乙酸乙酯):V(正己烷)=1:2)分离得到0.89 g白色固体,收率为51%。mp 199.0~201.5 ℃。1H NMR(600 MHz, CDCl3), δ:8.22(d, J=8.62 Hz, 1H), 8.10(d, J=8.07 Hz, 1H), 7.81(s, 1H), 7.78(d, J=7.70 Hz, 1H), 7.58(ddd, J=1.10, 7.20, 8.39 Hz, 1H), 7.36~7.40(m, 1H), 7.28~7.35(m, 2H), 7.07(d, J=3.30 Hz, 1H), 6.47(d, J=3.48 Hz, 1H), 4.74(d, J=4.77 Hz, 2H), 2.35(s, 3H), 2.14(br s, 1H); 13C NMR(151 MHz, CDCl3), δ:155.6, 146.9, 143.2, 141.4, 139.6, 137.3, 135.0, 129.5, 129.1, 127.8, 124.7, 124.6, 123.4, 122.2, 113.4, 111.4, 109.8, 57.6, 21.3。MS(ESI)计算值C19H16N2O4S [M+H]+:369.1, 实测值:369.1。
将1.00 g(4.72 mmol) 3-(5′-甲醛-2′-呋喃)-吲唑(中间体6)溶于5 mL DCM, 加入1.21 g (7.08 mmol)溴化苄,缓慢滴入1 mL三乙胺,室温搅拌6 h。反应后,加水、二氯甲烷萃取2次,有机层用饱和食盐水萃取2次后,减压旋蒸。经硅胶层析柱(V(甲醇):V(DCM)=1:2)分离得到1.22 g白色固体。将1.22 g(4.04 mmol)白色固体在干燥的环境下,溶于无水乙醇,在冰浴下搅拌5 min,缓慢加入0.46 g(12.12 mmol)硼氢化钠, 持续冰浴搅拌10 min后恢复室温,搅拌3 h。充分反应后,加入稀盐酸溶液淬灭,调pH值至7,抽滤除去沉淀,减压旋蒸。经硅胶层析柱(V(乙酸乙酯):V(正己烷)=1:3)分离得到1.01 g白色固体,收率为71%。mp 111.0~112.5 ℃。1H NMR(600 MHz, CDCl3), δ:8.05(d, J=8.07 Hz, 1H), 7.33~7.37(m, 1H), 7.29 (dd, J=7.98, 14.76 Hz, 3H), 7.23~7.26(m, 1H), 7.19~7.23(m, 3H), 6.87(d, J=3.30 Hz, 1H), 6.46(d, J=3.30 Hz, 1H), 5.64(s, 2H), 4.74(s, 2H), 3.47(s, 1H)。MS(ESI)计算值C19H16N2O2[M+H]+:305.1, 实测值:305.1。
MTT法:将处于对数生长期的HepG2细胞接种于96孔板(5000个/孔),培养24 h后给药,药物浓度梯度设置为0.1、1、10及20 μmol/L,给药48 h后进行MTT活性检测。
细胞增殖实验:将处于对数生长期的HepG2细胞接种于12孔板(0.5×105个/孔),培养24 h后给药,药物浓度梯度设置为1、10及20 μmol/L,分别于给药1、2和3 d后进行细胞计数。
蛋白质印迹法(Western Blot assay):将处于对数生长期的HepG2细胞接种于60 mm培养皿,培养24 h后,按照1、10和20 μmol/L的浓度梯度给药,同时予以低氧模拟剂去铁胺(Desferrioxamine, DFX)130 μmol/L处理细胞,给药24 h后收集细胞,裂解细胞制备蛋白样品,经8%的十二烷基硫酸钠聚丙烯酰胺凝胶电泳(SDS-PAGE)、转膜、封闭后,分别以1:1000/1:5000的HIF-1α/β-actin(β-肌动蛋白)抗体4 ℃孵育过夜,经1:5000的二抗室温孵育1 h,应用ECL试剂盒显影。
荧光定量逆转录聚合酶链反应(Real time-PCR):将处于对数生长期的HepG2细胞接种于35 mm培养皿,培养24 h后,按照1、10和20 μmol/L的浓度梯度给药,同时予以DFX 130 μmol/L处理细胞,给药24 h后收集细胞,提取核糖核酸(RNA),逆转录为互补DNA(cDNA),应用ABI公司生产的stepONE实时定量PCR仪检测VEGF信使RNA(mRNA)的水平。
目标化合物的合成见Scheme 1和Scheme 2。在Scheme 1中首先将吲唑(化合物1)与碘晶体溶解于DMF中,在碱性条件下生成3-碘吲唑,将其与1-Boc-4-甲烷磺酰氧基哌啶在碳酸钾的催化下生成中间体2[9-10]。中间体2与5-甲醛基呋喃-2-硼酸溶解于DME中,在饱和碳酸氢钠中加入催化剂四(三苯基膦)钯,经微波反应后用稀盐酸处理获得中间体3[11]。中间体3经硼氢化钠催化的还原反应,获得3-(5′-羟甲基-2′-呋喃)-1-哌啶基吲唑(化合物4)。化合物4经简单的脱水缩合反应可获得化合物5a~5d。
基于本课题组已报道的二芳醚类抗肿瘤活性的研究结果[7-8],本文设计并合成出了9个1,3-取代吲唑衍生物4、5a~5d、7a~7d。采用MTT法测试化合物对人肝细胞癌HepG2细胞株的体外增殖抑制作用[12]。实验重复3次,取平均值,阳性对照药物为7d(YC-1)(表 1)。在此基础上进一步比较了不同浓度的化合物7b与阳性对照YC-1对HepG2细胞存活率的抑制作用,采用细胞计数检测了化合物7b对HepG2细胞体外增殖的抑制作用(图 2);应用Western blot及Real time-PCR等方法检测了化合物7b对HIF-1及其下游基因VEGF表达的靶向抑制作用(图 3)。
 下载:
导出CSV
下载:
导出CSV
 |
|||
| Compounds | R | IRb@20 μmol/L/%a | IC50/(μmol·L-1) |
| 4 |  |
7.93 | not testedb |
| 5a |  |
8.83 | not testedb |
| 5b |  |
61.01 | 12.41 |
| 5c |  |
19.33 | not testedb |
| 5d |  |
16.34 | not testedb |
| 7a |  |
36.18 | not testedb |
| 7b |  |
62.57 | 10.37 |
| 7c |  |
45.83 | not testedb |
| 7d(YC-1) |  |
54.21 | 16.56 |
| a.IRb:Irbesartan; b.not tested as the inhabitation rate of preliminary screening less than 50%. | |||
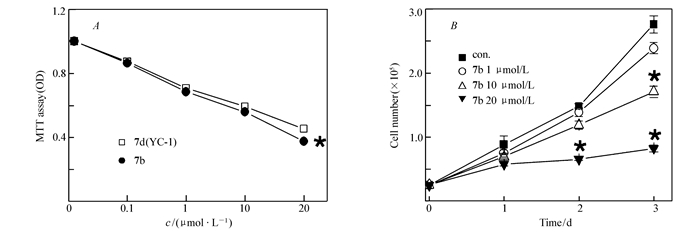
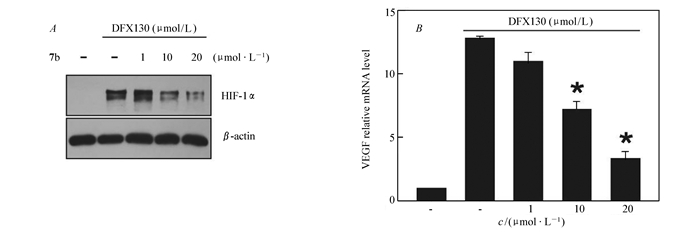
初步的细胞凋亡粗筛MTT试验后,我们可以从粗筛结果中看出一些变化:相较于具有活泼氮氢的化合物4,引入简单烷基结构(化合物5a~5d)能够提高化合物的HepG2细胞抑制率;化合物5a~5d对比可以看出简单烷基链并不是越长越有利,过长的或是空间结构更加复杂的烷基结构反而会降低化合物的抑制活性,很有可能是因为过多的烷基破坏了化合物原本空间的电子云分布,降低了其与对应受体的结合率;化合物7a~7c与已知化合物YC-1(化合物7d)结构相近,磺酰基的替换并没有对化合物的活性造成太大影响,甚至略有降低,但在苯环上引入甲基后有所改变,对位甲基取代的稳定结构提升了化合物的活性,很可能是因为甲基的存在形成共轭,使化合物的电子云密度分布发生改变(共平面化、趋于平均)。我们以50%为分界线,抑制率低于50%的化合物不再做进一步分析,而抑制率高于50%的化合物5b、7b和7d进行了进一步MTT分析求出其IC50值。实验结果表明,相较于阳性对照YC-1的抑制率(IC50=16.56 μmol/L)而言,化合物5b和7b表现出较好的抗肝癌活性,其中化合物7b的IC50=10.37 μmol/L尤为突出。苯环上引入推电子基团甲基后活性增加,其对位取代活性最强。初步推测可能与取代基的诱导效应及苯环上电子云密度有关。从图 2A可以看出,在给药化合物7b后,HepG2细胞存活率随着给药浓度增加逐渐降低,而且化合物7b对HepG2细胞的抑制率要优于阳性对照药YC-1。从图 2B可以看出,相对于空白对照,给药化合物7b后,HepG2细胞体外增殖速度显著降低,而且随着给药浓度增加,HepG2细胞体外增殖速度逐渐降低。化合物7b通过Western Blot及Real time-PCR等方法检测了其对HIF-1及其靶基因VEGF表达水平的影响,从图 3A可以看出,化合物7b可显著抑制HIF-1的蛋白表达,图 3B显示的是随着化合物7b给药浓度增加,HIF-1下游靶基因VEGF的mRNA表达水平逐渐被抑制。综上所述,新结构3-取代-1-对甲苯磺酰基吲唑类化合物具有靶向抑制HIF及良好的抗肝癌活性作用。由于化合物数量有限,因此其它取代基对抗肿瘤活性的影响有待于进一步研究。
本文设计合成了9个1, 3-取代吲唑衍生物。生物活性评价结果表明其具有不同程度的HIF-1抑制作用。将先导化合物YC-1的苄基结构替换为哌啶或苯磺酰结构,发现化合物5b和7b活性优于YC-1的化合物,为今后的设计提供了理论和实验依据。特别是化合物7b具有良好的HIF-1靶向抑制作用及抗肝癌活性(IC50=10.37 μmol/L),并较好地抑制了VEGF的表达水平。因此,化合物7b可作为潜在的抗肿瘤药物。更深层次机制,如HIF-1相关的信号通路和蛋白的关系,有待进一步研究。
梁宏元, 卢再鸣. 原发性肝癌综合介入治疗现状与困惑[J]. 临床肝胆病杂志, 2016,32,(1): 44-48. LIANG Hongyuan, LU Zaiming. Current Situation and Perplexity of Comprehensive Interventional Therapy for Primary Liver Cancer[J]. J Clin Hepatol, 2016, 32(1): 44-48.
Denko N C. Hypoxia, HIF1 and Glucose Metabolism in the Solid Tumour[J]. Nat Rev Cancer, 2008, 8(9): 705-713. doi: 10.1038/nrc2468
Tsui L, Fong T H, Wang I J. YC-1 Targeting of Hypoxia-Inducible Factor-1Α Reduces RGC-5 Cell Viability and Inhibits Cell Proliferation[J]. Mol Vis, 2012, 18: 1594-1603.
Tsui L, Fong T H, Wang I J. The Effect of 3-(5-Hydroxymethyl-2'-Furyl)-1-Benzylindazole(YC-1) on Cell Viability Under Hypoxia[J]. Mol Vis, 2013, 19: 2260-2273.
Lobb K, Hipskind P, Aikins J. Acyl Sulfonamide anti-Proliferatives:Benzene Substituent Structure-Activity Relationships for a Novel Class of Antitumor Agents[J]. J Med Chem, 2004, 47(22): 5367-5380. doi: 10.1021/jm030594r
刘菲, 刘楠, 何菱. 磺酰脒衍生物的合成及其抗肿瘤活性[J]. 合成化学, 2014,22,(4): 440-443. LIU Fei, LIU Nan, HE Ling. Synthesis and Antitumor Activities of Sulfonyl Amidine Derivatives[J]. Chinese J Synth Chem, 2014, 22(4): 440-443.
Yang S M, Huang Z N, Zhou Z S. Structure-based Design, Structure-Activity Relationship Analysis, and Antitumor Activity of Diaryl Ether Derivatives[J]. Arch Pharm Res, 2015, 38(10): 1761-1773. doi: 10.1007/s12272-015-0578-7
Hou J, Zhao W, Huang Z N. Evaluation of Novel N-(Piperidin-4-Yl) Benzamide Derivativesas Potential Cell Cycle Inhibitors in HepG2 Cells[J]. Chem Biol Drug Des, 2015, 86(2): 223-231. doi: 10.1111/cbdd.12484
Xiao J, Jin C, Liu Z. The Design, Synthesis, and Biological Evaluation of Novel YC-1 Derivatives as Potent Anti-Hepatic Fibrosis Agents[J]. Org Biomol Chem, 2015, 13(26): 7257-7264. doi: 10.1039/C5OB00710K
Zhang D, Kohlman D, Krushinski J. Design, Synthesis and Evaluation of Bicyclic Benzamides as Novel 5-HT 1F Receptor Agonists[J]. Bioorg Med Chem Lett, 2004, 14(24): 6011-6016. doi: 10.1016/j.bmcl.2004.09.079
An H, Kim N J, Jung J W. Design, Synthesis and Insight into the Structure Activity Relationship of 1, 3-Disubstituted Indazoles as Novel HIF-1 Inhibitors[J]. Bioorg Med Chem Lett, 2011, 21(21): 6297-6300. doi: 10.1016/j.bmcl.2011.08.120
Gamet-Payrastre L, Li P, Lumeau S. Sulforaphane, a Naturally Occurring Isothiocyanate, Induces Cell Cycle Arrest and Apoptosis in HT29 Human Colon Cancer Cells[J]. Cancer Res, 2000, 60(5): 1426-1433.

图 2 化合物7b显著抑制HepG2细胞的增殖
Figure 2 Proliferation of HepG2 cells was significantly inhibited by compound 7b
A.MTT assay was performed by YC-1 and different does compound 7b(n=4)(*P < 0.05); B.the growth curves of HepG2 cells treatment with different does compound 7b(n=3)(*P < 0.01)

图 3 化合物7b抑制HepG2细胞HIF-1α(A)及靶基因VEGF(B)的表达
Figure 3 Expression of HIF-1α and target gene VEGF were inhibited by compound 7b in HepG2 cells
A.western blot analysis was used to examine the expression of HIF-1α and β-actin proteins in HepG2 cells after incubation with compound 7b and DFX for 24 h; B.real time-PCR was used to examine the mRNA level of VEGF in HepG2 cells after incubation with compound 7b and DFX for 24 h(n=3)(*P < 0.01)
表 1 化合物4、5a~5d和7a~7d对HepG2细胞株的IC50
Table 1. IC50 values of compounds 4, 5a~5d, 7a~7d on HepG2 cell line in vitro
|
|
|||
| Compounds | R | IRb@20 μmol/L/%a | IC50/(μmol·L-1) |
| 4 | |
7.93 | not testedb |
| 5a | |
8.83 | not testedb |
| 5b | |
61.01 | 12.41 |
| 5c | |
19.33 | not testedb |
| 5d | |
16.34 | not testedb |
| 7a | |
36.18 | not testedb |
| 7b | |
62.57 | 10.37 |
| 7c | |
45.83 | not testedb |
| 7d(YC-1) | |
54.21 | 16.56 |
| a.IRb:Irbesartan; b.not tested as the inhabitation rate of preliminary screening less than 50%. | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: