Received Date:
02 November 2025 Revised Date:
16 April 2026 Available Online:
10 June 2026
Abstract:
Two density functional theory methods were employed to evaluate the H2 storage capabilities of metallo-borospherenes TM8B6 (TM=Ni, Pd). Consequently, the superatoms Ni8B6 and Pd8B6, which accommodate 40 and 32 H2 molecules, respectively, exhibit gravimetric H2 uptake capacities of 13.134% and 6.562%, respectively. The average binding energies of Ni8B6(H2)40 and Pd8B6(H2)32 fall within the optimal range for reversible H2 storage applications. The interactions between H2 molecules and the parent structures were characterized using various wave function analysis methods. Polarization effects, alongside the Kubas mechanism, are pivotal to the adsorption of H2 on TM8B6. Moreover, the investigations examine the effect of temperature on the H2 storage capacity of TM8B6 at atmospheric pressure. Atom-centered density-matrix propagation molecular dynamics simulations confirm the reversibility of H2 adsorption and desorption cycles. The thermodynamic analyses of the desorption behavior of H2 molecules were conducted via a three-dimensional graph, plotted based on the relationship between the number of adsorbed H2 molecules and temperature as well as pressure, revealing that the majority of adsorbed H2 molecules can be released at 0.5 MPa and 358 K. Compared to the respective monomeric counterparts, the H2 storage densities of (TM8B6)2 dimers exhibit a slight reduction.
As a result of CO2 emissions and fossil fuel depletion, a sustainable and renewable energy resource is urgently needed for transportation and industrial use to replace coal and oil[1]. Hydrogen energy has attracted worldwide attention due to its abundance, zero-polluting nature, renewability, and higher energy density compared to gasoline[2-3]. However, the implementation of H2 in transportation has faced challenges, primarily due to the absence of efficient hydrogen storage materials[4-5]. Traditionally, hydrogen gas is stored through low-temperature liquefaction and high-pressure compression[6-7]. However, these storage methods are not cost-effective because the processes of liquefaction and compression of H2 consume a substantial amount of energy. Furthermore, the installation of liquid and compressed gas hydrogen tanks on vehicles presents significant challenges due to their cumbersome nature and the difficulties associated with securing them properly.
An optimal H2 storage medium must satisfy several criteria, including but not limited to high gravimetric density, recyclability, and cost-effectiveness[8-10]. As requested, the U.S. Department of Energy (US-DOE) has set specific targets for optimal materials intended for practical H2 storage applications. These targets include a H2 storage density of 5.5% for onboard vehicle H2 storage systems by the year 2025, as well as operational conditions ranging from 1 to 100 MPa and temperatures between 233 and 358 K for vehicles in operation[11]. Given all this, solid-state H2 storage media have emerged as a crucial research area due to their cost-effectiveness, resulting in the development of various forms like chemical hydrides[12-14], covalent organic frameworks (COFs)[15-16], metal-organic frameworks (MOFs)[17-19], and carbon nanostructures[20-21].
In chemical hydrides, dehydrogenation is slow and requires high temperatures because H2 molecules dissociate into atoms, forming covalent bonds with the host material. For example, although magnesium hydride (MgH2) has a H2 mass ratio of 7.6%, its slow reaction kinetics hinder H2 desorption at ambient temperature[22-23]. In contrast, COFs, MOFs, and pure carbon materials exhibit low adsorption enthalpies, rendering them chemically inert and thus impractical for effective H2 storage under ambient conditions. To address the issue, previous studies indicated that MOFs[24-25], COFs[26-29], and carbon-based materials, including fullerene[30-34], nanotubes[35-38], and graphene[39-41], exhibit a substantial enhancement in hydrogen storage capacity when decorated with transition metal (TM) atoms. In all these cases, H2 adsorption occurs around the TM through orbital hybridization or polarization mechanisms. Consequently, a relatively strong interaction was established between the metal and hydrogen, resulting in an increased storage capacity and elevated desorption temperature. For example, Wei et al.[27] showed that in the modified COF-1 structure, each Sc atom can tightly adsorb four H2 molecules, achieving a H2 uptake of 6.78%. Yildirim et al.[30] employed the first-principles calculation to demonstrate that light TM-decorated C60 had significant H2 storage capability. Han et al.[37] reported that the nickel-decorated carbon nanotubes demonstrated a significant H2 uptake capacity of 0.87% at 298 K and 100 MPa, representing an enhancement factor of 2.5 compared to the untreated carbon nanotubes. However, the aggregation of TM atoms has been identified as a significant obstacle, mainly due to their extremely high binding energy, which in turn leads to a significant decrease in H2 storage capacity[42]. To address the aggregation problem, some researchers turned their attention from carbon-based materials to boron-based ones. Meng et al.[43] observed that, in contrast to their behavior on carbon nanostructures, titanium atoms on boron nanotubes tend to remain isolated from one another. After this, various TM-decorated boron-based materials, such as boron fullerene[44-49], boron nanotubes[50], borane[51-53], and boron clusters[54-58], have been the subjects of extensive research in the context of H2 storage. For example, Tang et al. have explored scandium-doped all-boron cages, specifically B40[44] and B28[45], as promising candidates for H2 storage materials. Additionally, Dong et al.[46] reported that a Ti-doped B40 cluster (Ti6B40) can adsorb up to 34 H2 molecules, achieving a maximum weight density of 8.70%. Sun et al.[49] employed DFT methods to evaluate the H2 storage potential of the highly stable La3B18 cluster, in which each lanthanum (La) atom could bind up to 6 H2 molecules, thereby achieving a 5.6% weight density. Zhao et al.[50] identified a low-energy single-walled scandium triboride (ScB3) nanotube that is capable of adsorbing approximately 6.1% H2, with a binding energy ranging from 22 to 26 kJ·mol-1. The Chattaraj group reported that B6H6Sc, B6H6Ti, and B6H6V complexes could adsorb up to 4 H2 molecules, achieving gravimetric H2 uptakes of 6.51%, 6.36%, and 6.21%, respectively[52].
Most recently, the smallest perfect cubic metallo-borospherenes TM8B6 (TM=Pd, Ni) have been identified as superatoms, adhering to the 18-electron rule and exhibiting spherical aromaticity and enhanced stability[59]. As far as we know, the H2 storage capacities of TM8B6 have not been studied. As previously discussed, boron-based complexes often exhibit exceptional H2 storage capabilities. Consequently, in the present study, the H2 storage performances of TM8B6 clusters were systematically investigated.
1.
Computational methods
The structures of isolated and hydrogenated TM8B6 were optimized without symmetry constraints using the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional[60-61], in conjunction with Grimme′s empirical dispersion correction (D3)[62]. In comparison to the other functionals, the PBE method demonstrates greater suitability for investigating hydrogen adsorption in TM compounds[33, 63-64]. In computational simulations, Pd was represented using the Stuttgart-Dresden double-ζ (SDD) basis set, whereas other atoms were modeled with the 6-311G(d, p) basis set. In the calculation of harmonic frequencies, there are no imaginary frequencies, thereby confirming the minimum energy structures. The average adsorption energy (ΔEa) and continuous adsorption energy (ΔEs) per H2 molecule were calculated to evaluate the adsorption capability and the reversibility of hydrogen storage of TM8B6 clusters, respectively. They are defined as follows:
where $ {E}_{{\rm{T}}{{\rm{M}}}_{8}{{\rm{B}}}_{6}} $, $ {E}_{{{\rm{H}}}_{2}} $, $ {E}_{{\rm{T}}{{\rm{M}}}_{8}{{\rm{B}}}_{6}({{\rm{H}}}_{2}{)}_{n}} $, $ {E}_{{\rm{T}}{{\rm{M}}}_{8}{{\rm{B}}}_{6}({{\rm{H}}}_{2}{)}_{n-8}} $ are the total energies of TM8B6, H2, TM8B6(H2)n, and TM8B6(H2)n-8, respectively. Of particular note is that if ΔEs > 0, H2 can be continuously adsorbed. Conversely, ΔEs < 0 inhibits further adsorption. The full counterpoise method was employed to correct for basis set superposition error (BSSE) in the calculation of adsorption energy[65]. To assess the reliability of the employed method, we have recalculated certain adsorption energies utilizing the ωB97XD density functional in conjunction with the same basis set. The ωB97XD functional, known for accurately predicting non-covalent interactions, was used to calculate adsorption energies with long-range interactions considered[66]. Comparable results were obtained at both computational levels; therefore, unless otherwise specified, the subsequent discussion will focus on the PBE(D3) level.
To further investigate the interaction mechanisms between atoms in the isolated and hydrogenated TM8B6, the quantum theory of atoms in molecules (QTAIM)[67] was applied. The molecular electrostatic potential (ESP) and atomic dipole-corrected Hirshfeld atomic charge (ADCH)[68] distributions were carried out to analyze the electronic structures of the free and hydrogen-adsorbed TM8B6. Localized orbital locator (LOL)[69], electron localization function (ELF) analyses,[70-72] and projected density of states (PDOS) analyses were performed to reveal the bonding nature. All calculations were facilitated by the Multiwfn software[73]. Additionally, the study employed the independent gradient model (IGM) based on the Hirshfeld partition of molecular density (IGMH)[74] to elucidate the bonding characteristics of the hydrogenated TM8B6 clusters. The thermodynamic stabilities of TM8B6 compounds were examined using Born-Oppenheimer molecular dynamics (BOMD) simulations. Hydrogen adsorption and desorption cycles were analyzed for reversibility using atom-centered density matrix propagation (ADMP)[75], which can offer comparable insights to BOMD while requiring fewer computational resources.
All the DFT calculations mentioned were implemented using the Gaussian 09 code[76].
2.
Results and discussion
2.1
Geometrical structures of metallo-borospherene TM8B6
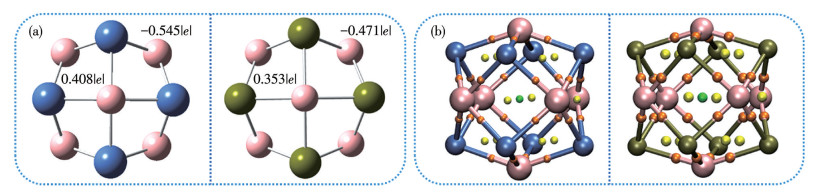
The most stable configurations of TM8B6 clusters have a low-lying Oh geometry, as illustrated in Fig.1a. All the bond distances of isolated and hydrogenated Ni8B6 and are Pd8B6 listed in Table 1 and 2, respectively. The calculated TM—B bond lengths for Ni8B6 and Pd8B6 are 0.189 and 0.209 nm as reported (0.191 and 0.207 nm) by Ao et al.[59], respectively. Furthermore, the thermodynamic stabilities of TM8B6 were evaluated through BOMD simulations conducted at 1 000 K, employing the PBE/6-31G(d) level of theory. The original structures utilized for the simulations are based on the optimized configurations at the PBE(D3)/6-311G(d, p) level of theory. A simulation duration of 3 ps is implemented with a trajectory time step of 0.5 fs. As illustrated in Fig.S1 (Supporting information), the relative potential energies of Ni8B6 and Pd8B6 exhibit minor oscillation around a consistent value throughout the simulation period. Analysis of the extractive snapshots (inset in Fig.S1) of TM8B6 at various simulation intervals (0, 1 500, and 3 000 fs) reveal that TM8B6 can consistently retain its respective cage-like structure, indicating a high degree of dynamic stability. The notable stabilities of the TM8B6 configurations can be ascribed to their unique structural and bonding characteristics. Take Ni8B6 as an example, the filled highest occupied molecular orbital (HOMO)-14 (1S2), triply degenerate HOMO-9 ($ {P}_{x}^{2} $, $ {P}_{y}^{2} $, and $ {P}_{z}^{2} $), doubly degenerate HOMO-1 (eg) ($ {D}_{{x}^{2}-{y}^{2}}^{2} $and $ {D}_{{z}^{2}}^{2} $) and triply degenerate HOMO (t2g) ($ {D}_{xz}^{2} $, $ {D}_{yz}^{2} $, and $ {D}_{xy}^{2} $) depicted in Fig.S2 correspond to the electronic configuration (1S21P61D10), indicating that Ni8B6 can be regarded as a superatom in accordance with the 18-electron rule, a classification that aligns well with prior research findings[59]. As shown in Fig.S3, an analogous analysis indicat that Pd8B6 can be considered a superatom according to the 18-electron rule. To further elucidate the nature of the high stability of TM8B6, QTAIM analyses were conducted. Fig.1b illustrates the molecular graphs of TM8B6 featuring bond critical points (BCPs), ring critical points (RCPs), and cage critical points (CCPs).
Figure 1
Figure 1.
(a) Optimized structures with ADCH charge of Ni8B6 (left) and Pd8B6 (right); (b) Molecular graphs of Ni8B6 (left) and Pd8B6 (right)
Pink, cyan, tan, orange, yellow, and green balls are B, Ni, Pd, BCP, RCP, and CCP, respectively.
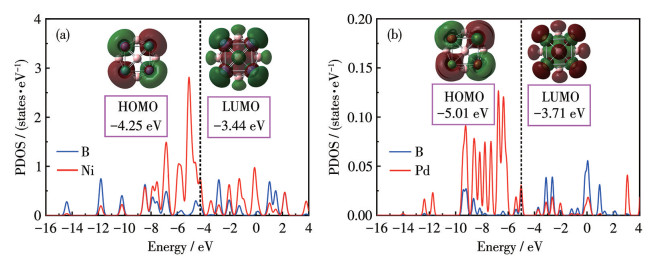
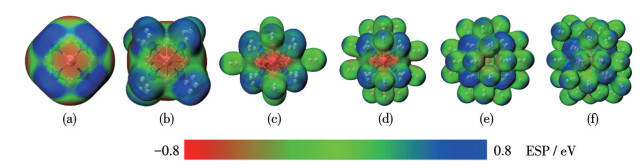
Table 3 presents the key to the pological parameters at BCPs for TM8B6, including electron density (ρ(r)), Laplacian of electron density (∇2ρ(r)), and total energy density (H(r)). In the TM8B6 clusters, BCPs are observed for TM—B interactions, while bond paths and BCPs between TM atoms are not present, indicating that TM atoms do not aggregate. Based on topological criteria, the interaction with ρ(r) at BCPs greater than 0.2 a.u., and H(r) less than 0 a.u. is typically considered a covalent bond[77]. As demonstrated in Table 3, Ni—B and Pd—B interactions, characterized by low ρ(r) (0.119 and 0.097 0 a.u., respectively) and negative H(r) values, predominantly exhibit covalent characters. For comparison, we additionally computed the ELF and LOL values at BCPs. If ELF and LOL values are greater than 0.5, it signifies regions of localized electrons, including both non-bonding and bonding electrons, while ELF and LOL values below 0.5 denote regions of delocalized electrons[66]. As presented in Table 2, the values of ELF and LOL exhibit a high degree of consistency. More importantly, the bond characteristics of TM—B, as calculated by ELF and LOL, exhibites strong concordance with the findings from the topological analyses. The interactions were further examined by analyzing the PDOS shown in Fig.2. The notable hybridization of Nid and Pdd orbitals with Bp orbitals, spanning from -15 to -4 eV and -12 to -4 eV, respectively, indicates that the presence of strong covalent bonds between TM and B atoms. As illustrated in Fig.1a, the ADCH charge analysis reveals that B and Ni atoms in Ni8B6 possess charges of approximately -0.545|e| and 0.408|e|, respectively. Similarly, B and Pd atoms in Pd8B6 carried about -0.471|e| and 0.353|e|, respectively. These observations indicate substantial electron transfer from TM atoms to the adjacent B atoms. Consequently, the TM atoms are induced into a cationic state, suggesting the potential for H2 adsorption is via a polarization mechanism. Furthermore, an ESP examination of the Ni8B6/Pd8B6 cluster is depicted in Fig.3a and Fig.S5a. The corresponding color scales are provided at the bottom of the figures. It is clearly shown that Ni/Pd atoms exhibit positive potentials (indicated in blue), while B atoms display negative potentials (indicated in red). These distributions of ESP accord with the ADCH analysis, suggesting that H2 molecules are more likely to be preferentially adsorbed onto the Ni/Pd atoms.
Table 3
Table 3.
Principal topological data of the bare TM8B6 and the TM8B6(H2)n complexes
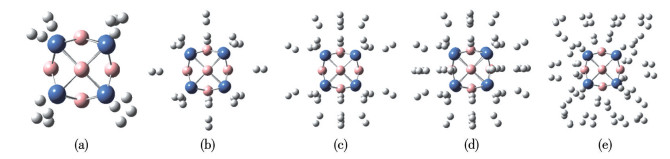
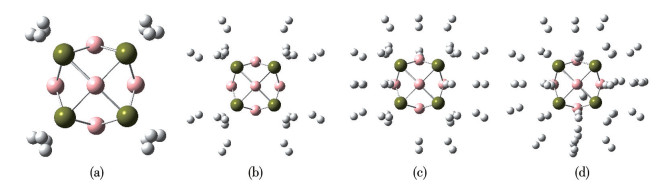
To survey the H2 storage capacity of metallo-borospherene TM8B6, multiple H2 molecules were incrementally introduced, followed by optimization of the host structures. This process was iteratively conducted until ΔEa decreased to below 0.10 eV, which ensured that H2 molecules were physisorbed, making it appropriate for reversible adsorption under operational conditions. The resulting structures of Ni8B6(H2)n and Pd8B6(H2)n are shown in Fig.4 and 5, respectively. The Ni8B6 and Pd8B6 clusters can bind up to 40 and 32 H2 molecules, respectively. The saturated complexes Ni8B6(H2)40 and Pd8B6(H2)32 have maximum H2 uptake densities of 13.134% and 6.562%, respectively, surpassing the US-DOE target of 5.5%. In Ni8B6(H2)40 and Pd8B6(H2)32, the initial 8 H2 molecules are adsorbed onto TM in a side-on configuration, with the H2 axis perpendicular to the line connecting the TM atoms and the H2 center. The axes of the subsequently adsorbed H2 molecules are vertical to the host. Table 1 summarizes the typical bond distances of the hydrogenated TM8B6. The hydrogens are incorporated into Ni8B6 and Pd8B6 clusters molecularly with H—H bond lengths ranging from 0.077 0 to 0.848 nm and 0.076 4 to 0.795 nm, respectively. In TM8B6(H2)8, the TM—B bond lengths exhibit a slight increase relative to those in free TM8B6 due to TM-H2 interactions. However, as additional H2 molecules are adsorbed by TM8B6(H2)8, the TM—B bond lengths are almost unchanged. With each step of sequential adsorption, H2 molecules are adsorbed farther from the TM sites due to steric hindrance. As presented in Table 1 and 2, the ΔEs values of TM8B6(H2)n are in the range of 0.013 0 to 0.680 eV by two methods, indicating that up to 40 and 32 H2 molecules can be effectively adsorbed onto Ni8B6 and Pd8B6, respectively. The predicted ΔEa for Ni8B6(H2)40 and Pd8B6(H2)32 is 0.207 and 0.103 eV at the PBE(D3) level, respectively, which are close to 0.206 and 0.110 eV calculated at the ωB97XD functional. Obviously, the TM8B6 clusters exhibit an adsorption strength between physisorption and chemisorption, making them potentially ideal for H2 storage under ambient conditions. It is well known that a large energy gap (ΔEgap) between HOMO and the lowest unoccupied molecular orbital (LUMO) can reflect the high chemical stability of a compound[52-53]. As shown in Fig.S4, the values of ΔEgap are calculated to analyze the stabilities of the isolated and hydrogenated TM8B6. The ΔEgap is different between the two methods; however, the tendency is generally consistent. It can be found that the ΔEgap values of the hydrogenated TM8B6 are larger than those of the corresponding bare cluster TM8B6. The analysis result shows that the cluster TM8B6 will be more stable after multiple H2 molecules are adsorbed. Furthermore, the computed ESP for the hydrogenated Ni8B6 and Pd8B6 is presented in Fig.3 and S5. The areas surrounding Ni/Pd atoms transition from blue to light blue with successive H2 adsorption, signifying the saturation of H2 uptake after the addition of 40 or 32 H2 molecules.
Figure 4
Figure 4.
View of Ni8B6 with (a) 8 H2, (b) 16 H2, (c) 24 H2, (d) 32 H2, and (e) 40 H2
The spheres colored gray, blue, and pink are H, Ni, and B atoms, respectively.
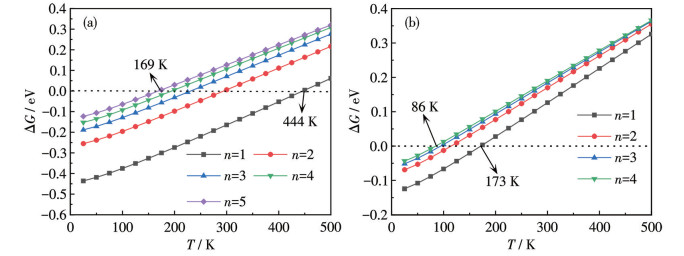
In the onboard application, it is essential to consider the influence of temperature and pressure on the processes of H2 adsorption and desorption. Consequently, we underscore the significance of applying Gibbs free energy corrections to achieve an accurate assessment of H2 adsorption. The Gibbs free energy change (ΔG) for the reaction TM8B6+8nH2→TM8B6(H2)8n (TM=Ni, n=1-5; TM=Pd, n=1-4) was examined to identify the favorable temperature range. The ΔG is defined as:
where $ {G}_{{\rm{T}}{{\rm{M}}}_{8}{{\rm{B}}}_{6}({{\rm{H}}}_{2}{)}_{n8}} $, $ {G}_{{\rm{T}}{{\rm{M}}}_{8}{{\rm{B}}}_{6}} $ and $ {G}_{{{\rm{H}}}_{2}} $ is the Gibbs free energies of TM8B6(H2)8n, TM8B6 and H2, respectively. According to the definition, ΔG < 0 suggests that the adsorption of H2 on TM8B6 occurs spontaneously. In contrast, ΔG > 0 signifies that the adsorption of H2 is energetically unfavorable. Fig.6a and 6b show the ΔG changes of Ni8B6(H2)8n and Pd8B6(H2)8n with temperature at 101.325 kPa, respectively. Obviously, ΔG exhibits a gradual increase as the temperature rises. For Ni8B6(H2)8n (n=1-5), all ΔG values are negative when the temperature is less than 169 K. Upon reaching 169 K, the adsorbed H2 molecules begin to relax. As the temperature further increased to ambient conditions, 32 H2 molecules are fully desorbed from Ni8B6(H2)40. It indicates that a H2 addition and release of 10.790% can be readily achieved under 101.325 kPa. For Pd8B6(H2)8n (n=1-4), all adsorbed H2 molecules start releasing from the host at 86 K and are completely desorbed by 173 K. This process allows for a 6.562% H2 uptake and release at atmospheric pressure.
Figure 6
Figure 6.
ΔG changed with temperature for (a) Ni8B6(H2)8n (n=1-5) and (b) Pd8B6(H2)8n (n=1-4) at 101.325 kPa
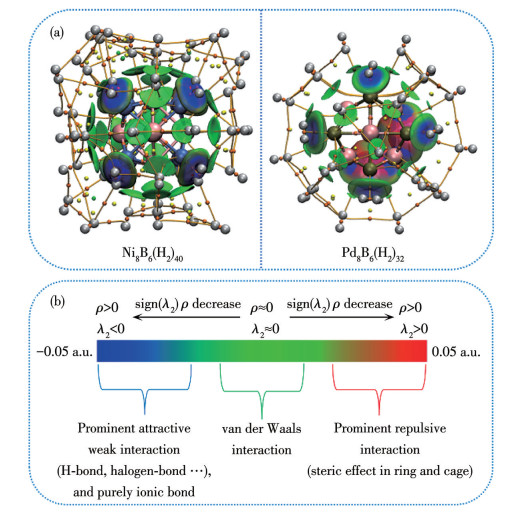
To investigate the interactions between atoms within hydrogenated TM8B6 clusters, topological analyses were conducted on saturated hydrogen-adsorbed clusters. The molecular graphs and topological parameters of Ni8B6(H2)40 and Pd8B6(H2)32 are detailed in Fig.7 and Table 3, respectively. The topological parameters associated with the B—TM bonds closely resemble those of the isolated TM8B6 motifs, suggesting that the structural integrities of the host clusters are preserved after the incorporation of H2 molecules. Furthermore, it is found that ELF and LOF values at the BCPs of H2 are approximately 1.000, suggesting that the absorbed H2 molecules retain their molecular forms. Additionally, it is noted that BCPs are formed between TM/B atoms and the H2 molecules. The B—H bonds are characterized as weak noncovalent interactions, as evidenced by low values of ρ(r), ELF, and LOF, along with positive ∇2ρ(r) and H(r) values. The interactions between TM and the initial 8 H2 molecules exhibit partial covalent characteristics, as evidenced by slightly elevated values of ρ(r), ∇2ρ(r), electron ELF and LOF, along with negative H(r) at BCPs. In contrast, the other BCPs between TM/B and H2 display low values of ρ(r), ∇2ρ(r), and H(r) approaching zero, indicating that these interactions are weak and noncovalent nature.
Figure 7
Figure 7.
(a) Molecular graphs of Ni8B6(H2)40 and Pd8B6(H2)32; (b) Standard color scale and chemical explanation of sign(λ2)ρ based on IGMH analysis[74]
a: Gray, pink, cyan, tan, orange, yellow, and green balls are H, B, Ni, Pd, BCP, RCP, and CCP, respectively; Inset: sign(λ2)ρ with IGMH δginter=0.005 a.u. isosurface.
The interaction characteristics between H2 and the host structures in Ni8B6(H2)40 and Pd8B6(H2)32 were further investigated through IGMH analysis. The isosurface maps of sign(λ2)ρ with IGMH δginter=0.005 a.u for Ni8B6(H2)40 and Pd8B6(H2)32 are presented in Fig.7a. Fig.7b depicts the IGMH color scale, in which blue, green, and red represent strong attractive interactions, weak van der Waals attractions, and steric hindrance, respectively. As shown in Fig.7a, the isosurface with a blend of blue and green hues suggests that the interactions between TM and the initial 8 H2 molecules exhibit an intermediate nature between van der Waals forces and strong attractive interactions.
Conversely, the BCPs between the remaining H2 molecules and the hosts are characterized predominantly by van der Waals interactions, as evidenced by the green isosurfaces. In prior research, exemplified by compounds Ti6B40[46] and La3B18[49], a single TM atom demonstrates the capacity to store up to 6 and 5 H2 molecules through Kubas interactions, respectively. However, in the cases of Ni8B6(H2)40 and Pd8B6(H2)32, each Ni or Pd atom binds only 1 H2 molecule via Kubas interaction[78], while the remaining H2 molecules interact with the host structures through van der Waals forces.
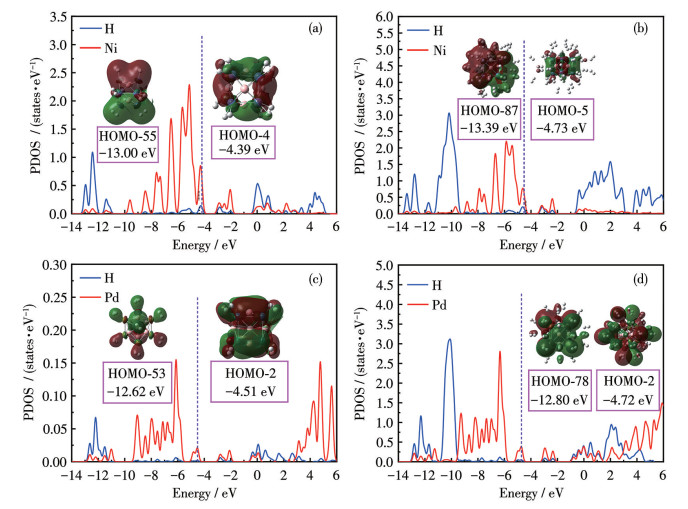
Fig. 8 presents the PDOS plots for the hydrogenated metallo-borospherene TM8B6. Fig. 8a illustrates that upon adsorption of an initial H2 molecule by each Ni atom, the overlap between σ orbitals of H2 molecules and the 3d orbitals of Ni occurs within the energy ranges of -13.00 to -11.37 eV, which are below the HOMO levels of Ni8B8(H2)8. Meanwhile, σ* orbitals of H2 molecules interact with the 3d orbitals of Ni. In brief, the interactions between H2 and Ni8B6 follow the Kubas mechanism. This facilitates a minor electron transfer from the σ orbitals of H2 to the 3d orbitals of Ni, accompanied by enhanced back-donation of electrons from the 3d orbitals of Ni to the σ* orbitals of H2. Consequently, the ADCH charge of each Ni atom increases from 0.408|e| to 0.472|e|. Furthermore, the bond lengths of the initially adsorbed H2 molecules are elongated relative to the subsequent 32 H2 molecules. As shown in Fig. 8b, when Ni8B8 reaches saturation adsorption, the overlap regions between σ orbitals of H2 molecules and the d orbitals of Ni expand to the energy ranges of -13.30 to -10.16 eV. This expansion suggests a decrease in the adsorption strength of H2 as more H2 molecules are adsorbed, a trend corroborated by the ΔEa values presented in Table 2. Likely, in Pd8B8(H2)8, σ orbitals of H2 molecules exhibit overlap with the d orbital of Pd within the energy range of -12.64 to -11.50 eV. Consequently, the charge on each Pd atom decreases from 0.353|e| to 0.288|e|. In contrast, this orbital overlap in Pd8B8(H2)32 extends over a broader energy range of -12.20 to -10.13 eV, along with the decrease in adsorption strength of H2.
Figure 8
Figure 8.
PDOS of (a) Ni8B6(H2)8, (b) Ni8B6(H2)40, (c) Pd8B6(H2)8, and (d) Pd8B6(H2)32
Dashed lines represent HOMO levels; Inset: partial occupied molecular orbitals of (a) Ni8B6(H2)8, (b) Ni8B6(H2)40, (c) Pd8B6(H2)8, and (d) Pd8B6(H2)32
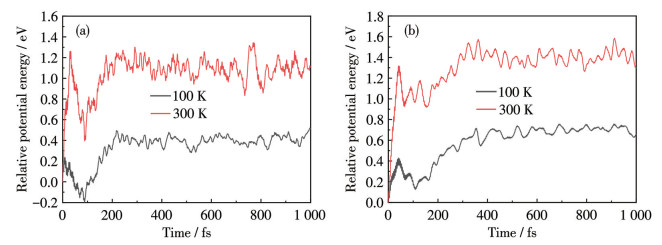
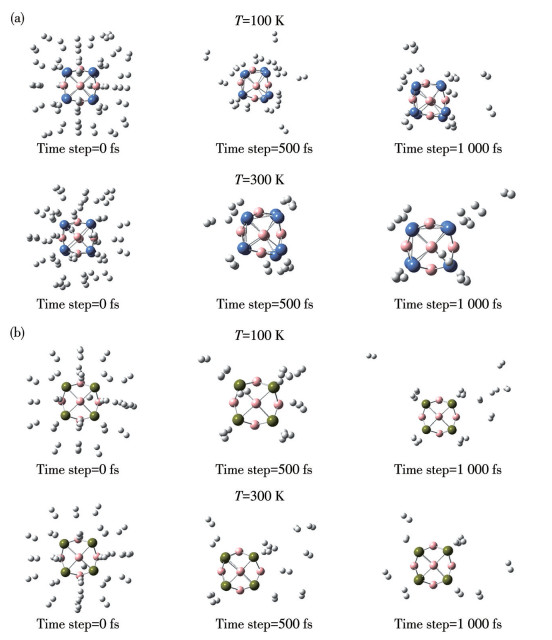
Next, we conducted ADMP molecular dynamics simulations to investigate the stability, reversibility, and desorption kinetics of saturated hydrogen-adsorbed complexes at varying temperatures and 101.325 kPa. Temperature regulation was exclusively achieved through the velocity scaling method. Saturated hydrogen-adsorbed clusters are subjected to 100 and 300 K at a 101.325 kPa for 1 000 fs with a 0.2 fs time step. Fig.9 and 10 illustrate the temporal trajectories of relative potential energies and snapshots from the ADMP molecular dynamics simulations, respectively. The average molecular motion increases with rising temperature, leading to the desorption of H2 molecules at shorter time intervals and at an accelerated rate. In the case of the Ni8B6(H2)40 cluster, 23 or 25 H2 molecules are released from the host simultaneously within time frames of 500 or 1 000 fs at a temperature of 100 K, respectively. In contrast, only 9 or 8 H2 molecules remain adsorbed during the same intervals at 300 K. For the Pd8B6(H2)32 clusters, 23 or 24 H2 molecules are expelled from the host within 500 or 1 000 fs at 100 K, respectively. Conversely, only 6 or 8 H2 molecules is adsorbed on the host during the same time intervals at 300 K. In short, the failure of the residual H2 molecules to completely desorb from the parent body is due to their ΔEa values being relatively high and approaching the range of chemical adsorption. This was consistent with the above calculation results. All the H2 molecules may leave the hosts at higher temperatures.
Figure 9
Figure 9.
Variation of relative potential energy of (a) Ni8B6(H2)40 and (b) Pd8B6(H2)32 vs simulation times at 100 and 300 K
For a material to be considered viable for H2 storage, it must be capable of adsorbing a substantial quantity of H2 molecules, while also allowing for their desorption under operational conditions. Consequently, a more pertinent parameter in the study of hydrogen storage may be the net difference between the number of H2 molecules adsorbed and desorbed. Therefore, to achieve a quantitative assessment of hydrogen adsorption and desorption over a broad range of temperature and pressure, we calculated the practical number of H2 molecules using thermodynamic analysis based on the grand canonical partition function[79].
where Z is the practical number of H2 molecules, n is the theoretical number of absorbed H2 molecules on the TM8B6, $ \Delta {E}_{{\rm{a}}}^{i} $ is the $ \Delta {E}_{{\rm{a}}} $ of the ith absorbed H2 molecule on the TM8B6, kB is the Boltzmann constant, and $ {\mu }_{{{\rm{H}}}_{2}} $ is the chemical potential of H2 at a specific temperature (T) and pressure (p):
where ΔH and ΔS denote the changes of enthalpy and entropy, respectively. The reference pressure (p0) was set at 0.1 MPa. The values for ΔH and ΔS are derived from the experimental database as cited in reference[80]. If N0* denotes the maximum number of adsorbed H2 molecules at 0 K, then Nave represents the average number of adsorbed H2 molecules at T and p, calculated as follows:
$ N_{{\rm{ave}}}=N_{0}*(Z-1)/Z(6) $
(6)
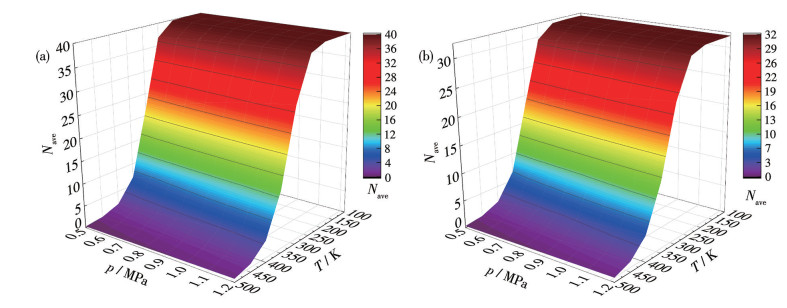
Based on equations 4-6, we present the Nave-p-T diagram in Fig.11, which quantitatively characterizes the H2 adsorption and desorption behavior of TM8B6 under p and T. Within the temperature range of 100-200 K and the pressure range of 0.5-1.2 MPa, Ni8B6 and Pd8B6 exhibit a maximum adsorption of 40 and 32 H2 molecules, respectively. As the temperature increased to 250 K, H2 molecules began to desorb from the host systems. Nevertheless, at 200 K within the pressure range of 0.5-1.2 MPa, the majority of H2 molecules remains adsorbed on the parent structures, with only 2-3 and 3-5 H2 molecules being released from Ni8B6 and Pd8B6, respectively. Upon further increasing the temperature to 500 K, complete desorption of H2 molecules occurs from both metallo-borospherene systems. Typically, for the practical process, the adsorption conditions are 1.2 MPa and 233 K, while for desorption conditions, they are 0.5 MPa and 358 K[11]. Table 4 presents the theoretical storage capacity (Gtheor) at 0 K, the number (Nads) of adsorbed H2 molecules under adsorption conditions, the number (Ndes) of adsorbed H2 molecules under desorption conditions, the practically usable number (Nprac=Nads-Ndes), and the effective storage capacity (Gprac). At the maximum theoretical capacity (Gtheor=13.134%) for Ni8B6, about 33 out of 40 H2 molecules are practically utilizable. Consequently, the practical gravimetric density decreases to 11.344%. In contrast, at the Gtheor=6.562% for Pd8B6, around 26 molecules can practically be useful out of 32 H2 molecules, leading to a practical gravimetric density of 5.412%. Therefore, the calculated Gprac of Ni8B6 and Pd8B6 are 11.344% and 5.412%, respectively, within the pressure range of 0.5-1.2 MPa and temperature range of 233-358 K.
Figure 11
Figure 11.Nave for (a) Ni8B6 and (b) Pd8B6 with change of temperature and pressure
2.5
Effects of TM8B6 oligomerization on H2 storage
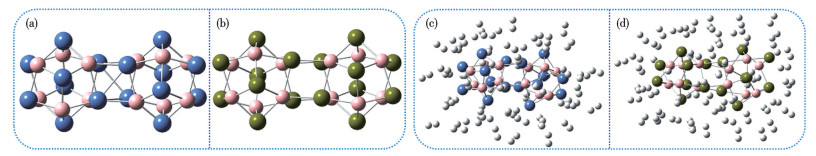
Although the aforesaid results are promising for the isolated TM8B6 clusters, several critical issues remain to be addressed. For instance, is it feasible to synthesize the cluster-assembled materials composed of TM8B6 clusters as building blocks while preserving the structural integrity of the individual TM8B6 clusters? To address these questions, we examined the interactions between two TM8B6 clusters in the dimer configuration TM16B12. The results of geometric structure optimization indicate that the two TM16B12 (Fig.12a, 12b) clusters can connect to each other via two staggered and parallel TM2B2 rings. The dimerization processes of TM16B12 are exothermic. To investigate the adsorption capacity of dimers for H2 molecules, we introduced 1 to 4 H2 molecules incrementally onto the TM atoms of TM16B12. During the optimization of Ni16B12(H2)64 and Pd16B12(H2)64, we find that 9 and 6 H2 molecules escape from the parent bodies, respectively. As illustrated in Fig.12c and 12d, the dimers Ni16B12 and Pd16B12 can accommodate a maximum of 55 and 58 H2 molecules, respectively, resulting in hydrogen weight densities of 9.412% and 5.983%. Although the adsorption capacities of the dimers (TM8B6)2 are slightly lower than those of the monomers (TM8B6), but they still surpass the set goal of 5.5%. Furthermore, the ΔEa values of Ni16B12(H2)55 and Pd16B12(H2)58 are 0.163 and 0.141 eV, respectively, suggesting their suitability for reversible H2 storage under ambient conditions.
Figure 12
Figure 12.
Optimized structures of (a) Ni16B12, (b) Pd16B12, (c) Ni16B12(H2)55, and (c) Pd16B12(H2)58
The spheres colored gray, blue, tan, and pink are H, Ni, Pd, and B atoms, respectively.
The metallo-borospherenes TM8B6 were investigated as potential H2 storage candidates by employing two DFT approaches. BOMD simulations reveal that these systems demonstrated excellent thermodynamic and dynamic stability at 1 000 K. The notable stabilities of TM8B6 can be attributed to the fact that they can be considered superatoms meeting the 18-electron rule. The analysis of PDOS reveal that the covalent interactions, arising from substantial overlaps between the TMd orbitals and the Bp orbitals, are predominant in TM8B6 clusters. Ni8B6 and Pd8B6 can at most attach 40 and 32 H2 molecules with a hydrogen uptake density of 13.134% and 6.562%, respectively, exceeding the target set by US-DOE. The ΔEa values for Ni8B6(H2)40/Pd8B6(H2)32 are 0.207/0.103 and 0.206/0.110 eV at the PBE(D3) and ωB97XD levels, respectively, satisfying the criteria for reversible H2 adsorption and desorption cycles under ambient conditions. The analyses of QTAIM, ELF, LOL, PDOS, and IGMH were employed to elucidate the interactions between H2 molecules and the parent structures, originating from electrostatic polarization and orbital hybridization. Thermos-chemistry calculations indicate that Ni8B6 and Pd8B6 have a H2 uptake and release of 10.790% and 6.562% at atmospheric pressure, respectively. ADMP molecular dynamics simulations revealed that the vast majority of H2 molecules in Ni8B6(H2)40 and Pd8B6(H2)32 can completely detach from the parent compounds within the temperature range of 100 to 300 K. The obtained practical storage densities of Ni8B6 and Pd8B6 are 11.344% and 5.412% at 0.5-1.2 MPa and 233-358 K, respectively. Despite potential reductions in H2 storage capacity, Ni16B12 and Pd16B12 dimers can effectively store 55 and 58 H2 molecules with a gravimetric density of 9.412% and 5.983%, respectively. The ΔEa values for Ni16B12(H2)55 and Pd16B12(H2)58 are determined to be 0.163 and 0.141 eV, respectively. These results indicate that the metallo-borospherenes TM8B6 exhibit potential as candidates for H2 storage. However, further experimental validation is necessary to substantiate these findings.
Acknowledgment:
This work was supported by Applied Research Program of Yuncheng University (Grant No.YY-202513).
Supporting information is available at http://www.wjhxxb.cn
[1]
HANLEY E S, DEANE J P, GALLACHÓIR B P Ó. The role of hydrogen in low carbon energy futures—A review of existing perspectives[J]. Renew. Sustain. Energy. Rev., 2018, 82(3): 3027-3045
[2]
SCHLAPBACH L, ZÜTTEL A, Hydrogen-storage materials for mobile applications[J]. Nature, 2001, 414: 353-358 doi: 10.1038/35104634
[3]
JENA P. Materials for hydrogen storage: Past, present, and future[J]. J. Phys. Chem. Lett., 2011, 2(3): 206-211 doi: 10.1021/jz1015372
[4]
MUHAMMED N S, GBADAMOSI A O, EPELLE E I, ABDULRASHEED A A, HAQ B, PATIL S, AI-SHEHRI D, KAMAL M S. Hydrogen production, transportation, utilization, and storage: Recent advances towards sustainable energy[J]. J. Energy Storage, 2023, 20: 109207
[5]
HIRSCHER M, YARTYS V A, BARICCO M, COLBE J B V, BLANCHAR D, BOWMAN R C. Materials for hydrogen-based energy storage—Past, recent progress and future outlook [J]. J. Alloy. Compd., 2020, 827(25): 153548
[6]
HU Y H. Novel hydrogen storage systems and materials[J]. Int. J. Energy Res., 2013, 37: 683-685 doi: 10.1002/er.3056
[7]
LUO W, CAMPBELL P G, ZAKHAROV L N, LIU S Y. A single-component liquid-phase hydrogen storage material[J]. J. Am. Chem. Soc., 2011, 133(48): 19326-19329 doi: 10.1021/ja208834v
[8]
KUBAS G J. Fundamentals of H2 binding and reactivity on transition metals underlying hydrogenase function and H2 production and storage[J]. Chem. Rev., 2007, 107(10): 4152-4205 doi: 10.1021/cr050197j
[9]
GRAETZ J. New approaches to hydrogen storage[J]. Chem. Soc. Rev., 2009, 38(1): 73-82 doi: 10.1039/B718842K
[10]
ZÜTTEL A, REMHOF A, BORGSCHULTE A, FRIEDRICHS O. Hydrogen: The future energy carrier[J]. Philos. Trans. R. Soc. A‒Math. Phys. Eng. Sci., 2010, 368(1923): 3329-3342
[11]
Hydrogen and Fuel Cell Technologies Office. DOE technical targets for onboard hydrogen storage for light-duty vehicles[EB/OL]. [2025-05-22]. https://www.energy.gov/eere/fuelcells/doe-technical-targets-onboard-hydrogen-storage-light-duty-vehicles
[12]
ORIMO S I, NAKAMORI Y, ELISEO J R, ZÜTTEL A, JENSEN C M. Complex hydrides for hydrogen storage[J]. Chem. Rev., 2007, 107(10): 4111-4132 doi: 10.1021/cr0501846
[13]
RUSMAN N A A, DAHARI M. A review on the current progress of metal hydrides material for solid-state hydrogen storage applications[J]. Int. J. Hydrog. Energy, 2016, 41(28): 12108-12126 doi: 10.1016/j.ijhydene.2016.05.244
[14]
KHAN H B, ZHANG T L. Metal hydride hydrogen storage risk assessment: A review[J]. J. Energy Storage, 2025, 129: 117273 doi: 10.1016/j.est.2025.117273
[15]
HAN S S, FURUKAWA H, YAGHI O M, GODDARD W A. Covalent organic frameworks as exceptional hydrogen storage materials[J]. J. Am. Chem. Soc., 2008, 130(35): 11580-11581 doi: 10.1021/ja803247y
[16]
FURUKAWA H, YAGHI O M. Storage of hydrogen, methane, and carbon dioxide in highly porous covalent organic frameworks for clean energy applications[J]. J. Am. Chem. Soc., 2009, 131(25): 8875-8883 doi: 10.1021/ja9015765
[17]
ROSI N L, ECKERT J, EDDAOUDI M, VODAK D T, KIM J, O′KEEFFE M, YAGHI O M. Hydrogen storage in microporous metal-organic frameworks[J]. Science, 2003, 300(5622): 1127-1129 doi: 10.1126/science.1083440
[18]
KAYE S S, DAILLY A, YAGHI O M, LONG J R. Impact of preparation and handling on the hydrogen storage properties of Zn4O(1, 4-benzenedicarboxylate)3 (MOF-5)[J]. J. Am. Chem. Soc., 2007, 129(46): 14176-14177 doi: 10.1021/ja076877g
[19]
AHMED A, SETH S, PUREWAL J, WONG-FOY A G, VEENSTRA M, MATZGER A J, SIEGEL D J. Exceptional hydrogen storage achieved by screening nearly half a million metal-organic frameworks[J]. Nat. Commun., 2019, 10: 1568-1576 doi: 10.1038/s41467-019-09365-w
[20]
DILLON A C, JONES K M, BEKKEDAHL T A, KIANG C H, BETHUNE D S, HEBEN M J. Storage of hydrogen in single-walled carbon nanotubes[J]. Nature, 1997, 386: 377-379 doi: 10.1038/386377a0
[21]
ZÜTTEL A, SUDAN P, MAURON P H, KIYOBAYASHI T, EMMENEGGER C H, SCHLAPBACH L. Hydrogen storage in carbon nanostructures[J]. Int. J. Hydrog. Energy, 2002, 27(2): 203-212 doi: 10.1016/S0360-3199(01)00108-2
[22]
IMAMURA H, MASANARI K, MUSUHARA M, KATSUMOTO H, SUMI T, SAKATA Y. High hydrogen storage capacity of nanosized magnesium synthesized by high energy ball-milling[J]. J. Alloy. Compd., 2005, 386(1/2): 211-216
[23]
PAIDAR V. Magnesium hydrides and their phase transitions[J]. Int. J. Hydrog. Energy, 2016, 41(23): 9769-9773 doi: 10.1016/j.ijhydene.2015.11.070
[24]
VILLAJOS J A, ORCAJO G, MARTOS C, BOTAS J Á, VILLACAÑAS J, CALLEJA G. Co/Ni mixed-metal sited MOF-74 material as hydrogen adsorbent[J]. Int. J. Hydrog. Energy, 2015, 40(15): 5346-5352 doi: 10.1016/j.ijhydene.2015.01.113
[25]
ZHAO D, WANG X X, YUE L L, HE Y B, CHEN B L. Porous metal-organic frameworks for hydrogen storage[J]. Chem. Commun., 2022, 58(79): 11059-11078 doi: 10.1039/D2CC04036K
[26]
DESHMUKH A, LE T N M, CHIU C C, KUO J L. DFT study on the H2 storage properties of Sc-decorated covalent organic frameworks based on adamantane units[J]. J. Phys. Chem. C, 2018, 122(29): 16853-16865 doi: 10.1021/acs.jpcc.8b06122
[27]
WEI S G, HUI Z, DAI J Q, CAI J M, YAN C X. First-principles study of hydrogen storage of Sc-modified semiconductor covalent organic framework-1[J]. ACS Omega, 2021, 6(34): 21985-21993 doi: 10.1021/acsomega.1c02452
[28]
ZHAO L, XU B Z, JIA J F, WU H S. A newly designed Sc-decorated covalent organic framework: A potential candidate for room-temperature hydrogen storage[J]. Comp. Mater. Sci., 2017, 137: 107-112 doi: 10.1016/j.commatsci.2017.05.017
[29]
PRAMUDYA Y, MENDOZA-CORTES J T. Design principles for high H2 storage using chelation of abundant transition metals in covalent organic frameworks for 0-700 bar at 298 K[J]. J. Am. Chem. Soc., 2016, 138(46): 15204-15213 doi: 10.1021/jacs.6b08803
[30]
YILDIRIM T, ÍÑIGUEZ J, CIRACI S. Molecular and dissociative adsorption of multiple hydrogen molecules on transition metal decorated C60[J]. Phys. Rev. B, 2005, 72: 153403 doi: 10.1103/PhysRevB.72.153403
[31]
ZHAO Y F, KIM Y H, DILLON A C, HEBEN M J, ZHANG S B. Hydrogen storage in novel organometallic buckyballs[J]. Phys. Rev. Lett., 2005, 94: 155504 doi: 10.1103/PhysRevLett.94.155504
[32]
SHIN W H, YANG S H, GODDARD W A, KANG J K. Ni-dispersed fullerenes: Hydrogen storage and desorption properties[J]. Appl. Phys. Lett., 2006, 88: 053111 doi: 10.1063/1.2168775
[33]
TIAN Z Y, DONG S L. Yttrium-dispersed C60 fullerenes as high-capacity hydrogen storage medium[J]. J. Chem. Phys., 2014, 140(8): 084706 doi: 10.1063/1.4866642
[34]
MAHAMIYA V, SHUKLA A, CHAKRABORTY B. Scandium decorated C24 fullerene as high capacity reversible hydrogen storage material: Insights from density functional theory simulations[J]. Appl. Surf. Sci., 2022, 573: 151389 doi: 10.1016/j.apsusc.2021.151389
[35]
YILDIRIM T, CIRACI S. Titanium-decorated carbon nanotubes as a potential high-capacity hydrogen storage medium[J]. Phys. Rev. Lett., 2005, 94: 175501 doi: 10.1103/PhysRevLett.94.175501
[36]
SINGH P, KULKARNI M V, GOKHALE S P, CHIKKALI S H, KULKARNI C V. Enhancing the hydrogen storage capacity of Pd-functionalized multi-walled carbon nanotubes[J]. Appl. Surf. Sci., 2012, 258: 3405-3409 doi: 10.1016/j.apsusc.2011.11.075
[37]
HAN Y J, PARK S J. Influence of nickel nanoparticles on hydrogen storage behaviors of MWCNTs[J]. Appl. Surf. Sci., 2017, 415: 85-89 doi: 10.1016/j.apsusc.2016.12.108
[38]
YANG L, YU L L, WEI H W, LI W Q, ZHOU X, TIAN W Q. Hydrogen storage of dual-Ti-doped single-walled carbon nanotubes[J]. Int. J. Hydrog. Energy, 2019, 44(5): 2960-2975 doi: 10.1016/j.ijhydene.2018.12.028
[39]
MASHOFF T, TAKAMURA M, TANABE S, HIBINO H, BELTRAM F, HEUN S. Hydrogen storage with titanium-functionalized graphene[J]. Appl. Phys. Lett., 2013, 103(1): 013903 doi: 10.1063/1.4812830
[40]
TANG C M, WAN Y M, ZHANG X, KANG J, ZOU J F, GAO J. The hydrogen storage properties of the Ti decorated benzene-Ti-graphene sandwich-type structures[J]. Int. J. Hydrog. Energy, 2016, 41(2): 1035-1043 doi: 10.1016/j.ijhydene.2015.12.014
[41]
YUAN L H, KANG L, CHEN Y H, WANG D B, GONG J J, WANG C N, ZHANG M L, WU X J. Hydrogen storage capacity on Ti-decorated porous graphene: First-principles investigation[J]. Appl. Surf. Sci., 2018, 434: 843-849 doi: 10.1016/j.apsusc.2017.10.231
[42]
SUN Q, WANG Q, JENA P, KAWAZOE Y. Clustering of Ti on a C60 surface and its effect on hydrogen storage[J]. J. Am. Chem. Soc., 2005, 127(42): 14582-14583 doi: 10.1021/ja0550125
[43]
MENG S, KAXIRAS E, ZHANG Z Y. Metal-diboride nanotubes as high-capacity hydrogen storage media[J]. Nano Lett., 2007, 7(3): 663-667 doi: 10.1021/nl062692g
[44]
TANG C M, ZHANG X. The hydrogen storage capacity of Sc atoms decorated porous boron fullerene B40: A DFT study[J]. Int. J. Hydrog. Energy, 2016, 41(38): 16992-16999 doi: 10.1016/j.ijhydene.2016.07.118
[45]
SI L, TANG C M. The reversible hydrogen storage abilities of metal Na (Li, K, Ca, Mg, Sc, Ti, Y) decorated all-boron cage B28[J]. Int. J. Hydrog. Energy, 2017, 42(26): 16611-16619 doi: 10.1016/j.ijhydene.2017.05.181
[46]
DONG H L, HOU T J, LEE S T, LI Y Y. New Ti-decorated B40 fullerene as a promising hydrogen storage material[J]. Sci Rep, 2015, 5: 09952 doi: 10.1038/srep09952
[47]
LIU P P, LIU F M, WANG Q M, MA Q. DFT simulation on hydrogen storage property over Sc decorated B38 fullerene[J]. Int. J. Hydrog. Energy, 2018, 43(42): 19540-19546 doi: 10.1016/j.ijhydene.2018.08.144
[48]
ESRAFILI M D, SADEGHI S. Y decorated all-boron B38 nanocluster for reversible molecular hydrogen storage: A first-principles investigation[J]. Int. J. Hydrog. Energy, 2022, 47(22): 11611-11621 doi: 10.1016/j.ijhydene.2022.01.160
[49]
SUN X Y, YIN P F, ZHANG Y, ZHANG C Y, FENG X, JIANG G. Efficient hydrogen storage capacity of La3B18: A DFT study[J]. Int. J. Hydrog. Energy, 2023, 48(21)7807-7813 doi: 10.1016/j.ijhydene.2022.11.261
[50]
ZHAO Y F, LUSK M T, DILLON A C, HEBEN M J, ZHANG S B. Boron-based organometallic nanostructures: Hydrogen storage properties and structure stability[J]. Nano Lett., 2008, 8(1): 157-161 doi: 10.1021/nl072321f
[51]
GUO C, WANG C. The hydrogen storage capacities of 4d transition metals in various boron systems[J]. J. Energy Storage, 2023, 57: 106216 doi: 10.1016/j.est.2022.106216
[52]
KONDAL R, KALAMSE V, DESHMUKH A, CHAUDHARI A. Closoborate-transition metal complexes for hydrogen storage[J], RSC Adv., 2015, 5(120): 99207-99216 doi: 10.1039/C5RA12927C
[53]
GUO C, WANG C. Stability and hydrogen storage properties of Mx-B6H6 complexes (M=Y-Mo, Ru-Ag, x=1-2)[J]. ACS. Sustain. Chem. Eng., 2021, 9(32): 10868-10881 doi: 10.1021/acssuschemeng.1c03363
[54]
马丽娟, 王剑锋, 贾建峰, 武海顺. B12Sc4和B12Ti4团簇的储氢性质[J]. 物理化学学报, 2012, 28(8): 1854-1860MA L J, WANG J F, JIA J F, WU H S. Hydrogen storage properties of B12Sc4 and B12Ti4 clusters[J]. Acta Phys.‒Chim. Sin., 2012, 28(8): 1854-1860
[55]
RODRÍGUEZ-KESSLER P L, RODRÍGUEZ-DOMÍNGUEZ A R, MACLEOD-CAREY D, MUÑOZ-CASTRO A. Exploring the size-dependent hydrogen storage property on Ti-doped Bn clusters by diatomic deposition: Temperature controlled H2 release[J]. Adv. Theory Simul., 2021, 4(7): 2100043 doi: 10.1002/adts.202100043
[56]
LIU P P, ZHANG Y F, XUA X J, LIU F M, LI J B. Ti decorated B8 as a potential hydrogen storage material: A DFT study with van der Waals corrections[J]. Chem. Phys. Lett., 2021, 765: 138277 doi: 10.1016/j.cplett.2020.138277
[57]
RAY S S, SAHOO R K, SAHU S. Reversible hydrogen storage in Ti decorated small boron clusters: Insights from molecular dynamics simulations[J]. J. Phys. Chem. Solids, 2023, 181: 111496 doi: 10.1016/j.jpcs.2023.111496
[58]
HUANG H S, LI G X, LI Z Q, ZHOU T Y, LI P, YANG X D, WU B. First-principles study of titanium-doped B7 cluster for high capacity hydrogen storage[J]. Molecules, 2024, 29(23): 5795 doi: 10.3390/molecules29235795
[59]
AO M Z, MA Y Y, MU Y M, LI S D. Perfect cubic metallo-borospherenes TM8B6 (TM=Ni, Pd, Pt) as superatoms following the 18-electron rule[J]. Nanoscale Adv., 2023, 5(23): 6688-6694 doi: 10.1039/D3NA00551H
[60]
PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Phys. Rev. Lett., 1996, 77: 3865 doi: 10.1103/PhysRevLett.77.3865
[61]
PERDEW J P, BURKE K, ERNZERHOF M. Errata: Generalized gradient approximation made simple[J]. Phys. Rev. Lett., 1997, 78: 1396
[62]
GRIME S, ANTONY J, EHRLICH S, KRIEG H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu[J]. J. Chem. Phys., 2010(132): 154104-154119
[63]
WADMERLAR N, KALAMSE V, CHAUDHARI A. Can ionization induce an enhancement of hydrogen storage in Ti2-C2H4 complexes?[J]. RSC Adv., 2012, 2(22)8497-8501 doi: 10.1039/c2ra21543h
[64]
GUO C, WANG C. Stability and hydrogen storage properties of Sc6O8 and Y6O8 cage-like complexes[J]. Int. J. Hydrog. Energy, 2023, 48(40): 15143-15153 doi: 10.1016/j.ijhydene.2022.12.325
[65]
BOYS S F, BERNARDI F. The calculation of small molecular interactions by the differences of separate total energies—Some procedures with reduced errors[J]. Mol. Phys., 1970, 19(4): 553-566 doi: 10.1080/00268977000101561
[66]
DU J G, SUN X Y, JIANG G, ZHANG C Y. The hydrogen storage on heptacoordinate carbon motif CTi72+[J]. Int. J. Hydrog. Energy, 2016, 41(26): 11301-11307 doi: 10.1016/j.ijhydene.2016.05.058
[67]
BADER R W F. Atoms in molecules: A quantum theory[M]. Oxford: Clarendon, 1990: 53-351
[68]
LU T, CHEN F W. Atomic dipole moment corrected Hirshfeld population method[J]. J. Theor. Comput. Chem., 2012, 11(1)163-183 doi: 10.1142/S0219633612500113
[69]
SCHMIDER H L, BECKE A D. Chemical content of the kinetic energy density[J]. Theochem-J. Mol. Struct., 2000, 527(1/2/3): 51-61
[70]
BECHKE A D, EDGECOMBE K E. A simple measure of electron localization in atomic and molecular systems[J]. J. Chem. Phys., 1990, 92(9): 5397-5403 doi: 10.1063/1.458517
[71]
SILVI B, SAVIN A. Classification of chemical bonds based on topological analysis of electron localization functions[J]. Nature, 1994, 371: 683-686 doi: 10.1038/371683a0
[72]
SAVIN A, NESPER R, WENGERT S, FÄSSLER T F. ELF: The electron localization function[J]. Angew. Chem.‒Int. Edit., 1997, 36(17): 1809-1832
[73]
LU T, CHEN F W. Multiwfn: A multifunctional wavefunction analyzer[J]. J. Comput. Chem., 2012, 33(5): 580-592 doi: 10.1002/jcc.22885
[74]
LU T, CHEN Q X. Independent gradient model based on Hirshfeld partition: A new method for visual study of interactions in chemical systems[J]. J. Comput. Chem., 2022, 43(8): 539-555 doi: 10.1002/jcc.26812
[75]
SCHELEGEL H B, IYENGAR S S, LI X S, MILLAM J M, VOTH G A, SCUSERIA G E, FRISCH M J. Ab initio molecular dynamics: Propagating the density matrix with Gaussian orbitals. Ⅲ. Comparison with Born-Oppenheimer dynamics[J]. J. Chem. Phys., 2002, 117(19): 8694-8704 doi: 10.1063/1.1514582
[76]
FRISCH M J, TRUCKS G W, SCHLEGEL H B, SCUSERIA G E, ROBB M A, CHEESEMAN J R, SCALMANI G, BARONE V, MENNUCCI B, PETERSSON G A, NAKATSUJI H, CARICATO M, LI X, HRATCHIAN H P, IZMAYLOV A F, BLOINO J, ZHENG G, SONNENBERG J L, HADA M, EHARA M, TOYOTA K, FUKUDA R, HASEGAWA J, ISHIDA M, NAKAJIMA T, HONDA Y, KITAO O, NAKAI H, VREVEN T, MONTGOMERY JR J A, PERALTA J E, OGLIARO F, BEARPARK M, HEYD J J, BROTHERS E, KUDIN K N, STAROVEROV V N, KOBAYASHI R, NORMAND J, RAGHAVACHARI K, RENDELL A, BURANT J C, IYENGAR S S, TOMASI J, COSSI M, REGA N, MILLAM J M, KLENE M, KNOX J E, CROSS J B, BAKKEN V, ADAMO C, JARAMILLO J, GOMPERTS R, STRATMANN R E, YAZYEV O, AUSTIN A J, CAMMI R, POMELLI C, OCHTERSKI J W, MARTIN R L, MOROKUMA K, ZAKRZEWSKI V G, VOTH G A, SALVADOR P, DANNENBERG J J, DAPPRICH S, DANIELS A D, FARKAS O, FORESMAN J B, ORTIZ J V, CIOSLOWSKI J, FOX D J. Gaussian 09, Revision D. 01[CP]. Gaussian, Inc., Wallingford, CT, 2009.
[77]
GREMER D, KRAKA E. Chemical bonds without bonding electron density—Does the difference electron-density analysis suffice for a description of the chemical bond?[J]. Angew. Chem.‒Int. Edit., 1984, 23(8): 627-628 doi: 10.1002/anie.198406271
[78]
KUBAS G. Metal-dihydrogen and σ-bond coordination: The consummate extension of the Dewar-Chatt-Duncanson model for metal-olefin π bonding[J]. J. Organomet. Chem., 2001, 635(1/2): 37-68
[79]
CALLEN H B. Thermodynamics and an introduction to thermostatistics[M]. 2nd ed. NewYork: Wiley, 1985.
[80]
LINSTROM P J, MALLARD W G. The NIST chemistry webbook: A chemical data resource on the internet[J]. J. Chem. Eng. Data, 2001, 46(5): 1059-1063 doi: 10.1021/je000236i
Figure 1
(a) Optimized structures with ADCH charge of Ni8B6 (left) and Pd8B6 (right); (b) Molecular graphs of Ni8B6 (left) and Pd8B6 (right)
Pink, cyan, tan, orange, yellow, and green balls are B, Ni, Pd, BCP, RCP, and CCP, respectively.
Figure 7
(a) Molecular graphs of Ni8B6(H2)40 and Pd8B6(H2)32; (b) Standard color scale and chemical explanation of sign(λ2)ρ based on IGMH analysis[74]
a: Gray, pink, cyan, tan, orange, yellow, and green balls are H, B, Ni, Pd, BCP, RCP, and CCP, respectively; Inset: sign(λ2)ρ with IGMH δginter=0.005 a.u. isosurface.

 下载:
下载:

 下载:
下载:













 下载:
下载:
