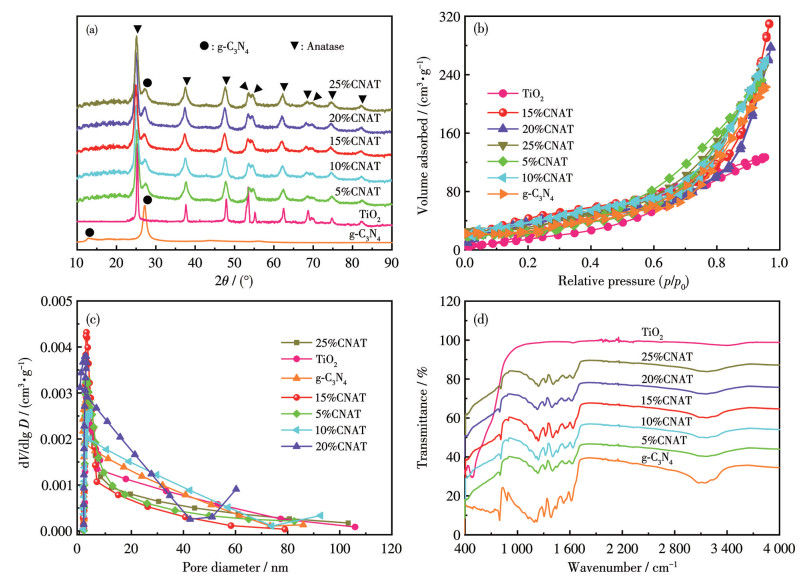
Figure 1.
(a) XRD patterns, (b) nitrogen adsorption-desorption isotherms, (c) corresponding pore-size distribution curves, and (d) FTIR spectra of g-C3N4, TiO2, and xCNAT
Construction of an S-scheme g-C3N4/TiO2 heterostructure for tetracycline degradation and hydrogen production
Mingze AN , Bingbing ZHANG , Zhao YANG , Hao PU , Weijie CHEN , Bin XUE , Sheng WANG , Xiaoyan DING , Lulu SHI

The acceleration of industrialization and the growing global demand for energy have placed water pollution control and clean energy development at the forefront of environmental and energy challenges[1]. TC antibiotics, widely used in medicine and agriculture, persist in aquatic environments as unmetabolized residues. Their release promotes bacterial resistance and bioaccumulation, threatening ecosystem stability and human health[2]. Conventional wastewater treatment methods often fail to eliminate these antibiotics and may cause secondary pollution. Meanwhile, hydrogen energy, characterized by its zero pollution and high energy density, is considered an ideal alternative to fossil fuels. Photocatalytic water splitting technology can directly harness solar energy to convert water into hydrogen, enabling clean energy production[3]. Therefore, developing materials capable of simultaneously achieving efficient pollutant degradation and hydrogen evolution represents a critical pathway for addressing both water contamination and energy shortages, bearing considerable academic value and practical significance.
Photocatalytic technology, recognized for its mild operating conditions, low energy consumption, and lack of secondary pollution, shows great potential in water purification and hydrogen production. Advancing this technology relies on developing high-performance photocatalysts. Titanium dioxide (TiO2), a well-established material, is widely used owing to its chemical stability, non-toxicity, and low cost[4]. However, its wide band gap limits light absorption largely to the ultraviolet region, and the rapid recombination of photogenerated carriers results in low efficiency, restricting practical applications. To overcome these drawbacks, strategies such as ion doping, noble metal deposition, and semiconductor coupling have been explored. Among them, constructing semiconductor heterojunctions has been particularly effective[5]. Coupling TiO2 with a narrow-band gap semiconductor not only broadens the spectral response but also promotes carrier separation via the built-in electric field at the interface, thereby enhancing photocatalytic activity.
Graphitic carbon nitride (g-C3N4), an emerging non-metallic semiconductor photocatalyst, possesses an appropriate band gap, remarkable visible-light response, high chemical stability, and low fabrication cost, making it an ideal partner for constructing heterojunctions with TiO2[6]. The band structure of g-C3N4 endows it with a strongly reductive conduction band for hydrogen evolution and a highly oxidative valence band for pollutant degradation. However, its practical application is limited by rapid charge recombination and a small specific surface area. By constructing a heterojunction with TiO2, the complementary properties of both materials are synergistically combined: the strong oxidation capability of TiO2 is integrated with the visible-light response of g-C3N4, overcoming their individual limitations and substantially enhancing the overall photocatalytic efficiency.
The band structure design of heterojunctions is crucial for photocatalytic performance. While conventional type-Ⅱ heterojunctions facilitate charge separation, they often compromise redox potential, failing to satisfy the stringent requirements for simultaneous pollutant degradation and hydrogen production. Recently, step-scheme (S-scheme) heterojunctions have garnered significant attention due to their unique charge transfer mechanism. By establishing appropriate band alignment, S-scheme heterojunctions promote the recombination of less reactive electrons and holes, while preserving those with strong redox power for photocatalytic reactions. This mechanism not only achieves efficient carrier separation but also maintains high redox activity[7]. Therefore, applying the S-scheme concept to the g-C3N4/TiO2 system provides an effective solution to the limited redox capacity of traditional heterojunctions, offering a promising strategy for enhancing performance in tetracycline (TC) degradation and hydrogen evolution.
Despite progress in g-C3N4/TiO2 heterojunctions, key challenges remain, including the unclear S-scheme formation mechanism, insufficient exploration of synergistic TC degradation and hydrogen production, and limited catalyst stability under realistic conditions. To address these issues, we constructed an S-scheme g-C3N4/TiO2 heterojunction via an integrated thermal polymerization-hydrothermal-calcination route. The composite was thoroughly characterized in terms of its structure, morphology, and optical properties. Its photocatalytic activity and stability were systematically evaluated for both pollutant degradation and hydrogen evolution, and the underlying S-scheme mechanism was elucidated. This work provides theoretical and experimental foundations for developing efficient and stable bifunctional photocatalysts, advancing the practical application of photocatalytic technology in environmental and energy fields.
All chemicals used were of analytical grade and are detailed in the Supporting Information.
g-C3N4 was synthesized by thermal polymerization. Specifically, a mixture of melamine (2.856 5 g), urea (1.283 4 g), and thiourea (0.860 1 g) was homogenized and transferred to a covered alumina crucible. The sample was heated in a tube furnace from room temperature to 550 ℃ at a ramp rate of 5 ℃·min-1 and held for 4 h. After cooling naturally to room temperature, the resulting light-yellow product was collected and gently ground into a fine powder.
For further processing, 0.2 g of the as-prepared powder was dispersed in 50 mL of absolute ethanol. The mixture was first stirred magnetically for 30 min, followed by ultrasonication for 5 h to achieve complete exfoliation. The resulting homogeneous suspension was dried at 60 ℃ in a forced convection oven. Finally, the dried product was gently ground to obtain the g-C3N4 photocatalyst.
The S-scheme g-C3N4/TiO2 heterojunction photocatalyst was prepared via a combined hydrothermal-calcination route. First, 0.3 g of g-C3N4 powder and 2 mL of glacial acetic acid were dissolved and dispersed in 100 mL of absolute ethanol under vigorous stirring. Then, 7.48 mL of tetrabutyl titanate was added dropwise into the dispersion, and stirring was continued for 1 h to allow preliminary hydrolysis. The resulting mixture was transferred to a Teflon-lined autoclave and hydrothermally treated at 180 ℃ for 12 h. After cooling naturally to room temperature, the precipitate was collected by centrifugation and washed repeatedly with ethanol and deionized water until the supernatant became neutral. The product was vacuum-dried at 80 ℃ overnight, gently ground, and finally calcined at 500 ℃ for 4 h (heating rate: 5 ℃·min-1) in a covered crucible. The resulting light-yellow powder was collected as the final photocatalyst (denoted as 15%CNAT, where 15% represents the mass fraction of g-C3N4 to tetrabutyl titanate).
To investigate the effect of g-C3N4 content on the photocatalytic activity of the S-scheme g-C3N4/TiO2 heterojunction, a series of samples with varying mass fractions (x) of g-C3N4 to tetrabutyl titanate (x=5%, 10%, 20%, and 25%) was synthesized. These catalysts, denoted as xCNAT, were prepared by following the aforementioned hydrothermal-calcination procedure while adjusting the mass ratios.
Furthermore, all experimental procedures for catalyst characterization and photocatalytic activity testing (including both pollutant degradation and hydrogen production) are detailed in the Supporting Information.
X-ray diffraction (XRD) analysis of g-C3N4, TiO2, and xCNAT confirmed their phase composition and crystal structure (Fig.1a). The XRD pattern of g-C3N4 exhibited two characteristic diffraction peaks at 2θ of 12.8° and 27.3°. The peak at 12.8° is assigned to the in-plane repeating tri-s-triazine units. In comparison, the intense peak at 27.3° corresponds to the (002) interlayer stacking of the conjugated aromatic system, representing a typical graphitic layered structure[8]. For TiO2, the diffraction peaks observed at 2θ of 25.3°, 37.8°, 47.9°, 53.8°, 54.9°, 62.5°, 68.6°, 70.1°, 74.9°, and 82.6° can be indexed to the (101), (004), (200), (105), (211), (204), (116), (220), (215), and (224) planes of the anatase phase (PDF No.21-1272)[9], confirming the high phase purity of the synthesized material without detectable impurities.
In the XRD patterns of xCNAT, the characteristic peaks of both g-C3N4 and anatase TiO2 were present, with no additional peaks corresponding to other phases. This confirms that the composite process did not induce any chemical reaction, allowing each component to retain its original crystal structure. The successful coexistence of these phases directly verifies the formation of a composite material. Furthermore, the absence of significant peak shifts compared to the components indicates unchanged lattice parameters and no induced lattice distortion at the heterojunction interface. Therefore, it can be concluded that the g-C3N4/TiO2 heterojunction is primarily formed through interfacial physical contact rather than lattice doping or phase transformation.
In summary, the XRD results verify the successful construction of a g-C3N4/TiO2 heterojunction with high crystallinity and preserved structural integrity in both components. This high degree of crystallinity helps minimize crystal defects and facilitates charge transport. Consequently, the established biphasic heterostructure, along with its inherent band alignment, is expected to induce an internal electric field, thereby creating a favorable foundation for efficient electron-hole separation and enhanced photocatalytic performance.
N2 adsorption-desorption measurements were performed to determine the textural properties of the samples (Fig.1b). All samples exhibited type Ⅳ isotherms with H3-type hysteresis loops above p/p0=0.4, indicative of mesoporous structures[10]. The Brunauer-Emmett-Teller (BET) surface area of TiO2 and g-C3N4 was 128 and 224 m2·g-1, respectively. Notably, the 15%CNAT composite exhibited the highest specific surface area of 311 m2·g-1, which is significantly higher than that of the individual components. This enhancement implies that the composite formation at this optimal ratio effectively modulates the textural properties, likely due to interfacial coupling that leads to a more developed porous framework. The pore size distribution analysis (Fig.1c) confirmed the predominance of mesopores (2-50 nm) in all samples and revealed that the 15%CNAT composite possessed the highest pore volume, aligning with its superior surface area. This optimal textural combination of high surface area and large mesopore volume creates a structure conducive to photocatalytic reactions: it provides abundant active sites and efficient transport channels for reactants and charge carriers[11], thereby establishing a foundation for enhanced performance.
Fourier transform infrared (FTIR) spectra were performed to analyze the chemical structures of TiO2, g-C3N4, and xCNAT over the wavenumber range of 400-4 000 cm-1 (Fig.1d). The spectrum of g-C3N4 showed characteristic bands at 500-1 500 cm-1 and 1 500-3 000 cm-1, corresponding to the stretching and bending vibrations of tri-s-triazine rings and the stretching modes of C—N/C=N bonds, respectively[12-13]. A broad band at 3 000-3 500 cm-1 is attributed to surface O—H and N—H groups[14]. In contrast, pure TiO2 displayed a broad absorption below 800 cm-1, characteristic of Ti—O—Ti vibrations in the anatase lattice[15]. The FTIR spectra of xCNAT retained the characteristic peaks of both g-C3N4 and TiO2 without detectable new bands. The persistence of triazine-related peaks and C—N/C=N vibrations from g-C3N4, while also clearly observing the Ti—O—Ti band of TiO2, indicates a physical combination without chemical reaction, further confirming the conclusions from XRD and N2 adsorption-desorption analyses.
To probe the surface chemical composition and electronic structure, both in-situ (with light) and ex-situ (without light) XPS characterization were conducted on g-C3N4, TiO2, and 15%CNAT. The survey spectra (Fig.2a) clearly showed characteristic peaks of C, N, Ti, and O. In particular, the C and N peaks originate from g-C3N4, while the Ti and O peaks are attributed to TiO2. The coexistence of all four elemental peaks in the 15%CNAT spectrum provides direct evidence for the successful formation of a composite between g-C3N4 and TiO2.
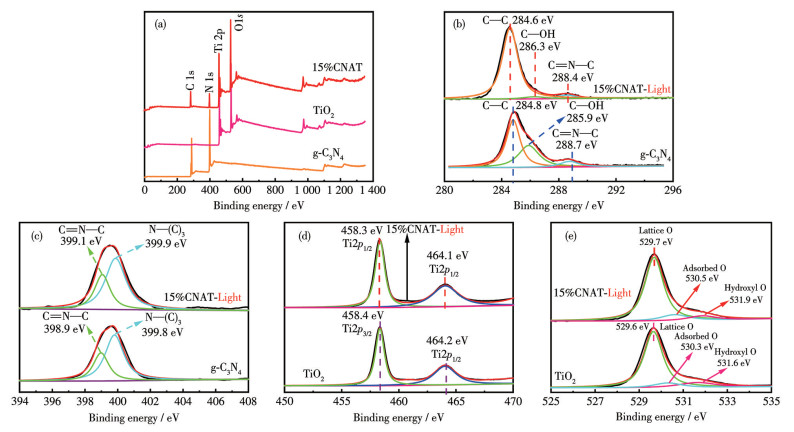
The deconvolution of the C1s XPS spectrum for g-C3N4 (Fig.2b) reveals three peaks at 284.8, 285.9, and 288.7 eV, which are ascribed to C—C, C—OH, and C=N—C bonds, respectively. This result confirms the presence of a graphitic heterocyclic structure[16]. In 15%CNAT, the characteristic peaks for C—C and C=N—C exhibited negative shifts of 0.2 and 0.3 eV in their binding energies, respectively, while the C—OH peak showed a positive shift of 0.4 eV. This indicates that the carbon skeleton of g-C3N4 remained intact and retained its fundamental bonding configuration.
The deconvoluted N1s spectrum of g-C3N4 (Fig.2c) displayed two fitted peaks located at 398.9 and 399.8 eV, corresponding to sp2 hybridized nitrogen in C=N—C bonds (within triazine rings) and tertiary nitrogen in N—(C)3 groups, respectively[17]. In the 15%CNAT composite, these peaks were observed at 399.1 and 399.9 eV. This represents a slight positive shift (0.2 eV) for the C=N—C species and a positive shift (0.1 eV) for the N—(C)3 group. The persistence of these characteristic nitrogen states indicates that the fundamental heterocyclic structure of g-C3N4 is largely maintained, while the observed shifts point to an interfacial interaction with TiO2 that does not disrupt the structural framework.
The Ti2p XPS spectrum of TiO2 (Fig.2d) showed two characteristic peaks at 458.4 eV (Ti2p3/2) and 464.2 eV (Ti2p1/2), as expected for Ti4+ [18]. In the 15%CNAT composite, both peaks shift slightly to lower binding energies of 458.3 and 464.1 eV, respectively. This negative shift implies electron transfer from g-C3N4 to TiO2, resulting in a modulated electronic structure that is favorable for enhanced separation of photogenerated charge carriers.
The deconvolution of the O1s spectrum for TiO2 (Fig.2e) reveals three components at 529.6, 530.3, and 531.6 eV, corresponding to lattice O, adsorbed O, and hydroxyl O, respectively[19]. In the composite, all three peaks exhibited positive shifts to 529.7, 530.5, and 531.9 eV. This consistent shift suggests an increased concentration of surface oxygen species, which is anticipated to supply more active sites for photocatalytic reactions.
To sum up, XPS analysis confirms the successful construction of 15%CNAT and reveals significant electronic interactions at the g-C3N4/TiO2 interface. These synergistic interactions preserve the intrinsic chemical structures of both components while effectively modulating the electronic state of Ti and the surface composition of O. The resultant favorable electronic and surface chemical environment is conducive to enhanced photocatalytic performance.
Fig.3 presents the scanning electron microscope (SEM), transmission electron microscope (TEM), high-resolution TEM (HRTEM), and selected area electron diffraction (SAED) results, which elucidate the microstructure and crystalline phase of g-C3N4, TiO2, and 15%CNAT. As shown in Fig.3a, g-C3N4 displayed a porous, layered morphology with uniformly distributed pores, offering a large specific surface area that facilitates molecular adsorption and transport. In comparison, TiO2 (Fig.3b) was composed of densely packed, homogeneously distributed nanoparticles, reflecting its typical particulate structure. Fig.3c and 3d demonstrated that 15%CNAT successfully combined the layered framework of g-C3N4 with the particulate features of TiO2. The TiO2 nanoparticles were uniformly anchored onto the g-C3N4 layers, forming a favorable structure for interfacial electron transfer and enhanced photocatalytic performance.

The TEM image in Fig.3e further corroborates the thin and porous layered structure of the composite. In Fig.3f, dark TiO2 nanoparticles were uniformly dispersed over the lighter g-C3N4 sheets with negligible agglomeration, indicating a high level of TiO2 distribution conducive to the exposure of active sites. The HRTEM image (Fig.3g) reveals two clear lattice fringe spacings of 0.32 and 0.35 nm, which match the (002) plane of g-C3N4[20] and the (101) plane of TiO2[21], respectively. This offers direct atomic-scale evidence for the successful integration of the two components, forming a well-defined and intimate interface. As shown in Fig.3h, the SAED pattern displayed diffraction rings corresponding to the (101) plane of TiO2 and the (002) plane of g-C3N4, in agreement with the HRTEM observations, thereby further confirming the coexistence and crystalline nature of both phases within the composite. Collectively, the electron microscopy analysis reveals that 15%CNAT maintained the porous layered framework of g-C3N4 and the nanocrystalline character of TiO2, the latter being uniformly and firmly anchored onto the sheets. Such a well-defined interface, achieved at both micro and atomic scales, provides a solid basis for the material′s superior photocatalytic performance.
The elemental composition and spatial distribution of 15%CNAT were investigated by an energy-dispersive spectrometer (EDS). Fig.4a presents a representative SEM image, while Fig.4b shows the corresponding overlay elemental mappings of C, N, O, and Ti. The uniform distribution of all four elements provides direct visual evidence for the homogeneous integration of g-C3N4 and TiO2 within the composite.
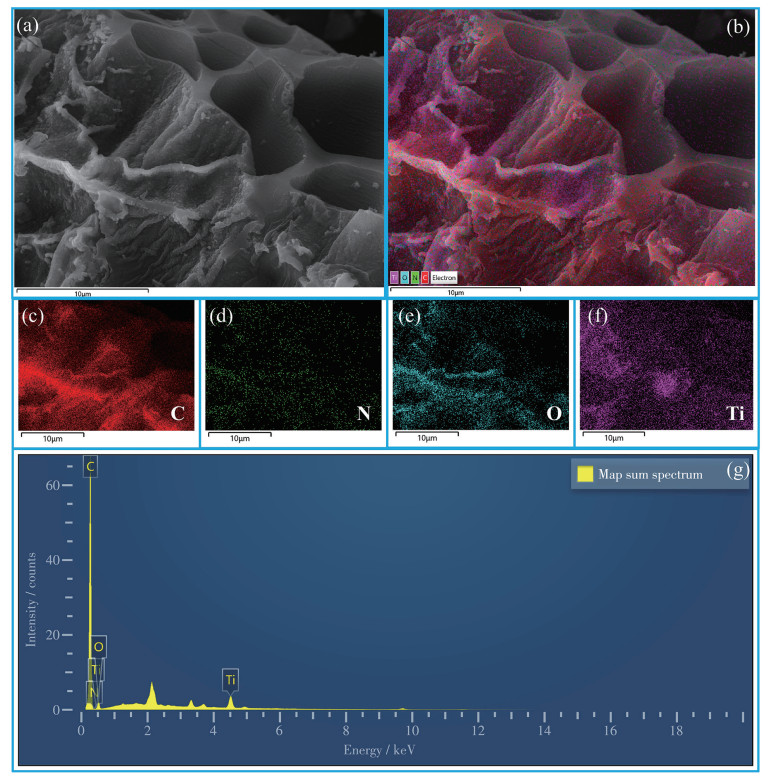
The corresponding elemental maps provide further structural insights: the C signal (Fig.4c) was continuous and uniform, consistent with the well-retained layered framework of g-C3N4. Similarly, the homogeneous distribution of N (Fig.4d) reflects the unagglomerated nitrogen-containing heterocyclic structure of g-C3N4. The widespread O signal (Fig.4e) originates from both the lattice oxygen in TiO2 and surface-adsorbed species. Finally, the Ti mapping (Fig.4f) showed finely dispersed particles, confirming the uniform deposition of TiO2 nanoparticles on the g-C3N4 sheets, in full agreement with the TEM and SEM results. The EDS survey spectrum (Fig.4g) clearly showed characteristic peaks of C, N, O, and Ti, confirming the expected chemical composition and the successful formation of the composite without impurities. In summary, EDS mapping definitively confirms the uniform distribution of g-C3N4 and TiO2 within 15%CNAT, verifying its successful formation.
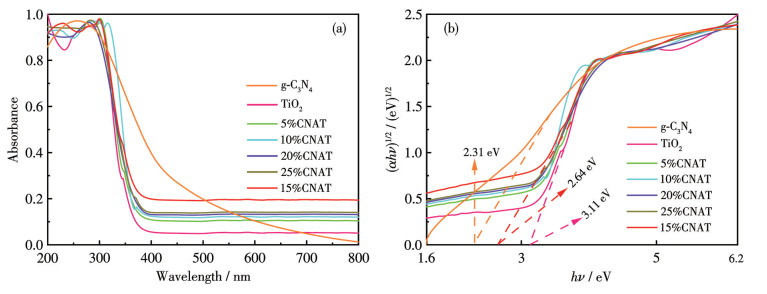
The optical absorption properties and band gap structures of g-C3N4, TiO2, and xCNAT were investigated using UV-visible diffuse reflectance spectra (UV-Vis DRS). The corresponding band gap energies were determined from the spectra via the Kubelka-Munk transformation method, as presented in Fig.5. As illustrated in Fig.5a, g-C3N4 exhibited strong absorption from 200 to 450 nm, which is attributed to the π-π* electron transitions characteristic of its visible-light response[22]. In contrast, TiO2 absorbed intensely only in the ultraviolet region (200-380 nm) due to its intrinsic band-to-band transition, which involves charge transfer from O2- to Ti4+ and is consistent with typical ultraviolet-responsive behavior[23]. xCNAT displayed a combined absorption profile, featuring enhanced ultraviolet absorption from TiO2 and preserved visible-light absorption from g-C3N4. Notably, 15%CNAT showed the most pronounced redshift in the absorption edge, implying an extended light-harvesting range and improved utilization of photon energy[24].
The band gap energies were estimated from the Kubelka-Munk plots (Fig.5b) by extrapolating the linear portion of the (αhν)1/2 versus hν curve. The band gap of g-C3N4 was determined to be 2.31 eV, characteristic of a visible-light-responsive semiconductor, while that of TiO2 was 3.11 eV, indicative of an ultraviolet-light-active material. Notably, 15%CNAT exhibited an intermediate band gap of 2.64 eV, indicating that the interfacial interaction between the two components effectively modulates the electronic structure. This tailored band gap strikes an effective balance, preserving the visible-light absorption of g-C3N4 while incorporating the strong ultraviolet-light response of TiO2, thereby broadening the composite′s overall light-harvesting range. These optimized optical properties promote more efficient solar energy utilization and significantly contribute to the improved photocatalytic performance.
The photocatalytic performance of the as-prepared samples was evaluated by the degradation of TC under simulated sunlight irradiation. Fig.6a shows the temporal variation of TC mass concentration with different catalysts. The reactions were carried out under the following conditions: illumination from a 300 W Xe lamp, catalyst dosage of 100 mg, initial TC mass concentration of 50 mg·L-1, pH=7, and temperature of 30 ℃. The degradation process was tracked using the relative concentration (ρt/ρ0).
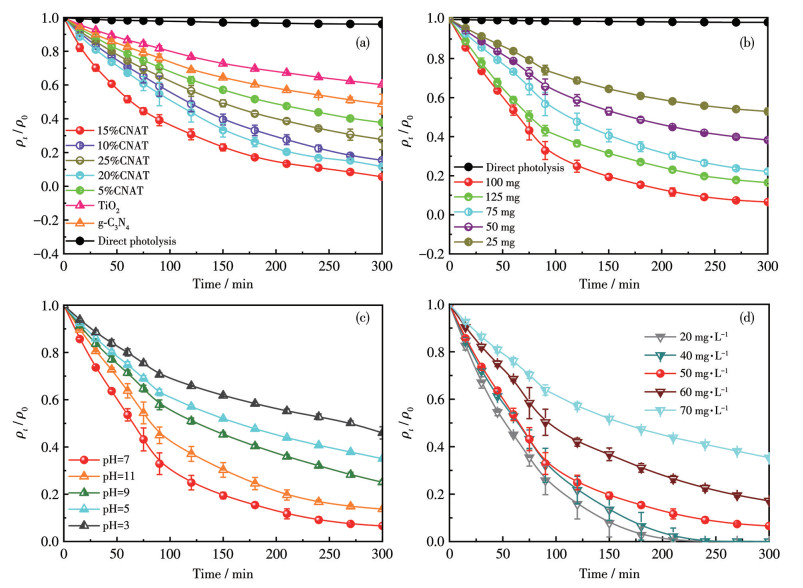
As shown in Fig.6a, TC exhibited negligible self-degradation in the absence of a catalyst, confirming that direct photolysis was insignificant. Both TiO2 and g-C3N4 displayed limited photocatalytic activity, with g-C3N4 performing slightly better than TiO2. This low efficiency is attributed to the rapid recombination of photogenerated electron-hole pairs in these single-component systems, which causes a gradual decrease in the degradation rate. In contrast, all xCNAT composites showed markedly enhanced TC degradation, with performance being composition-dependent. Notably, 15%CNAT achieved the most rapid decline in ρt/ρ0 within 300 min, indicating that its heterojunction structure was optimal for facilitating charge separation.
The photocatalytic degradation kinetics were further analyzed using a pseudo-first-order model, plotted as -ln(ρt/ρ0) versus reaction time (Fig.S1). The observed linear relationships validate that the process follows pseudo-first-order kinetics under the experimental conditions, which is consistent with the mass concentration trends in Fig.6a. The markedly higher reaction rate constant of 15%CNAT further corroborates that the S-scheme heterojunction between g-C3N4 and TiO2 effectively promotes the separation and transfer of photogenerated charges, thereby enhancing the photocatalytic degradation activity[25].
Fig.6b illustrates the effect of catalyst dosage on the photocatalytic degradation of TC. The experiments were conducted under fixed conditions: 300 W Xe lamp irradiation, an initial TC mass concentration of 50 mg·L-1, pH=7, temperature of 30 ℃, and catalyst loadings of 25, 50, 75, 100, and 125 mg, respectively. In the absence of a catalyst, the TC mass concentration remained nearly constant, confirming negligible photolysis and corroborating the findings in Fig.6a. The degradation rate was highly dependent on catalyst loading. When the dosage was 25 mg, the reaction was slow, with only a minor reduction in ρt/ρ0. As the dosage increased to 50 and 75 mg, the degradation rate rose substantially, evidenced by a steeper decline in ρt/ρ0 over 300 min. The optimal performance was achieved when the dosage was 100 mg, reaching a degradation efficiency of 94.5% after 300 min. However, further increasing the dosage to 125 mg results in a noticeable decline in performance. This trend can be explained by the fact that increasing the catalyst dosage initially supplies more active sites, thereby improving contact among TC molecules, photons, and the catalyst, thus enhancing degradation[26]. Conversely, an excessive dosage induces particle agglomeration, which diminishes the specific surface area, blocks active sites, and impedes light penetration, ultimately reducing photon utilization and reaction efficiency[27]. Hence, under the given conditions, the optimal dosage for the S-scheme heterojunction was 100 mg, which achieved an optimal balance between active site availability and light absorption, resulting in peak photocatalytic performance.
Fig.6c illustrates the effect of solution pH on the photocatalytic degradation of TC. The experiments were conducted under the following fixed conditions: 300 W Xe lamp irradiation, a catalyst dosage of 100 mg, an initial TC mass concentration of 50 mg·L-1, a temperature of 30 ℃, and pH values of 3, 5, 7, 9, and 11. The degradation efficiency of TC was highly dependent on pH. The optimal performance was observed at pH 7, with ρt/ρ0 approaching zero within 300 min, indicating nearly complete degradation. A moderately high degradation rate occurred at pH 11, followed by a slower rate at pH 9. In contrast, under acidic conditions, the efficiency decreased markedly, with the lowest degradation observed at pH 3. This pH-dependent behavior stems from the combined influence of pH on the molecular state of TC, the catalyst′s surface charge, and the formation of reactive species. At pH 7, the catalyst surface charge and the ionic form of TC are in favorable alignment, promoting adsorption and degradation. Furthermore, the generation and activity of photoinduced holes and hydroxyl radicals were optimized under these conditions[28]. In alkaline media, despite the reduced stability of certain radicals, reasonable degradation efficiency was still preserved. Under acidic conditions, however, TC protonation and an altered catalyst surface charge impede adsorption and diminish the yield of reactive oxygen species, resulting in significantly slower degradation[29]. In conclusion, pH 7 was identified as the optimal condition for TC degradation using the S-scheme g-C3N4/TiO2 heterojunction catalyst, as it ensures a synergistic balance among substrate-catalyst interaction, surface properties, and active species reactivity.
The photocatalytic degradation efficiency was investigated as a function of the initial TC mass concentration (20, 40, 50, 60, and 70 mg·L-1) under fixed conditions: 300 W Xe lamp irradiation, a catalyst dosage of 100 mg, pH 7, and a temperature of 30 ℃ (Fig.6d). The degradation rate exhibited a strong inverse correlation with the initial TC mass concentration. At a low mass concentration of 20 mg·L-1, rapid degradation was observed (ρt/ρ0≈0 within 210 min). Increasing the mass concentration to 40-50 mg·L-1 resulted in a gradual reduction in the degradation rate, although relatively high efficiency was maintained within 300 min. A further increase to 60-70 mg·L-1 led to a marked decline in performance. This trend is attributed to the availability of active sites and photon utilization efficiency. At low TC mass concentrations, the abundant active sites on the catalyst surface ensure sufficient contact between TC molecules and reactive species (e.g., photogenerated holes and ·OH), leading to rapid degradation[30]. As the mass concentration increased, the active sites became saturated. Concurrently, the higher TC mass concentration induces a shielding effect by absorbing incident photons, which reduces light penetration and lowers photon utilization. Furthermore, the accumulation of intermediate products can block active sites and further hinder the reaction. Consequently, the overall degradation rate declined with increasing TC mass concentration. These results indicated that the TC degradation efficiency by the S-scheme g-C3N4/TiO2 heterojunction decreased at higher initial mass concentrations. Therefore, for practical application, it is essential to consider both the initial pollutant mass concentration and the required removal target to optimize the operational parameters. Fig.S2 examines the influence of reaction temperature (10-50 ℃) on TC photocatalytic degradation, with other parameters fixed: 300 W Xe lamp irradiation, 100 mg catalyst, 50 mg·L-1 TC, and pH 7. Elevating the reaction temperature led to a noticeable increase in the TC degradation rate. At 30 ℃, the reaction proceeded rapidly with excellent efficiency. In contrast, at 10 ℃, degradation was slowest, showing only a marginal decrease in the ρt/ρ0 value over 300 min. The enhancement in degradation rate with temperature is due to improved reaction kinetics. Higher temperatures promote the migration of photogenerated charge carriers, thereby suppressing their recombination. Simultaneously, the collision frequency between active species (e.g., h+, and ·OH) and TC molecules increases, accelerating the degradation process. However, excessively high temperatures may compromise the catalyst′s crystal structure, reduce its stability, or lead to the premature decomposition of active intermediates[31]. Hence, for practical applications, the temperature must be optimized to balance reaction efficiency with the thermal stability of the catalyst. Collectively, the results demonstrate that strategically tuning the reaction temperature significantly enhances the TC degradation efficiency of the xCNAT catalysts, with 30 ℃ established as the optimum within the experimental range. This identifies temperature as a critical parameter and offers valuable guidance for scaling up the photocatalytic treatment of TC-containing wastewater.
Furthermore, the linearity of the plots of -ln(ρt/ρ0) versus time in Fig.S3-S6 indicates that the photocatalytic degradation of TC follows pseudo-first-order kinetics across all investigated conditions, including variations in catalyst dosage, solution pH, initial TC mass concentration, and reaction temperature.
The photocatalytic hydrogen evolution performance and cycling stability of each catalyst were evaluated via water splitting under simulated sunlight irradiation. As shown in Fig.S7, control experiments confirmed that no significant hydrogen evolution was detected in the absence of either the catalyst or light irradiation, ruling out contributions from non-photocatalytic processes. In contrast, only a trace amount of hydrogen was produced without triethanolamine (C6H15O3N, TEOA), attributable to the intrinsic but weak overall water-splitting activity of 15%CNAT. These results collectively demonstrate that the primary hydrogen evolution activity originates from the photocatalytic reaction of xCNAT.
As shown in Fig.7a, both TiO2 and g-C3N4 exhibited low H2 evolution rates. Although g-C3N4 showed a slight advantage over TiO2, their overall efficiency was limited by the rapid recombination of photogenerated charge carriers. In contrast, all xCNAT heterojunctions demonstrated markedly enhanced activity, with performance being composition-dependent. 15%CNAT achieved the highest H2 evolution rate of 329.1 μmol·h-1·g-1 over 8 h, underscoring its superior activity. This enhancement verifies the efficacy of the S-scheme heterojunction in promoting charge separation and migration, thereby facilitating the hydrogen evolution reaction. The cycling stability of 15%CNAT was assessed over 40 h under simulated sunlight, employing 100 mg of catalyst and 5 mL of TEOA as a sacrificial agent, as shown in Fig.7b. The hydrogen yield remained highly stable throughout the test, demonstrating robust photostability. This indicates that the S-scheme heterostructure effectively inhibits photocorrosion, thereby enhancing the catalyst′s durability and ensuring sustained long-term hydrogen production. As shown in the XRD pattern (Fig.S8), the used photocatalyst exhibited no detectable phase change.
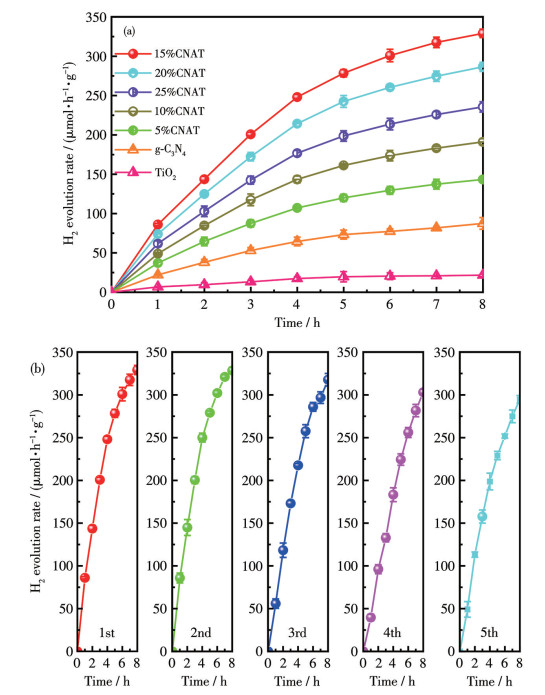
XPS analysis of the recovered 15%CNAT (Fig.S9) confirmed that the material retained its original C, N, O, and Ti composition without changes in the chemical states of these elements. Furthermore, no new peaks corresponding to strongly adsorbed impurities were detected. These findings collectively demonstrated the excellent structural stability of the catalyst, where the active sites remained undegraded (without oxidation or reduction), which is crucial for maintaining its catalytic activity.
The charge separation efficiencies of g-C3N4, TiO2, and 15%CNAT were investigated via transient photocurrent response and electrochemical impedance spectroscopy (EIS) under Xe lamp illumination. As shown in Fig.8a, all samples exhibited a reproducible photocurrent during on/off light cycles, confirming successful electron generation. 15%CNAT demonstrated a photocurrent density substantially greater than its individual constituents. Given that the photocurrent magnitude directly reflects charge separation efficiency, this enhancement signifies markedly suppressed carrier recombination in the heterojunction, which directly accounts for its superior photocatalytic performance[32].
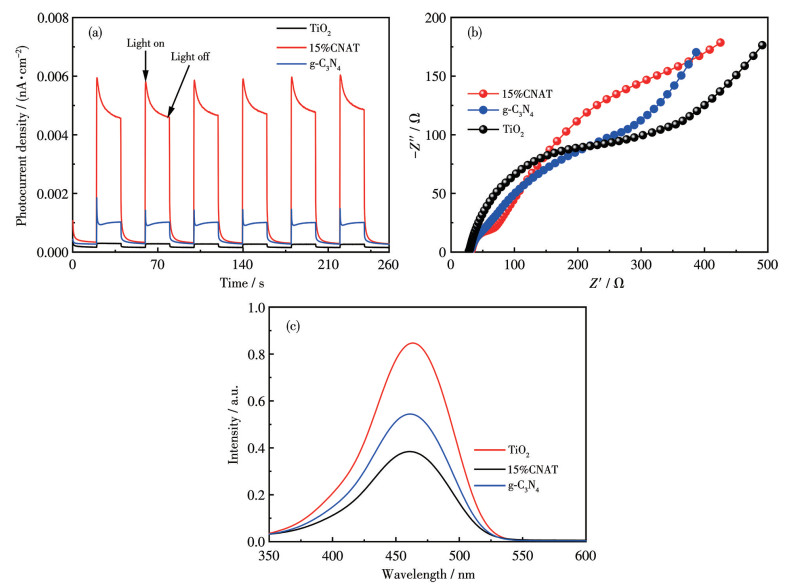
The EIS Nyquist plots in Fig.8b showed a markedly smaller arc radius for 15%CNAT compared to g-C3N4 or TiO2, indicating significantly reduced charge transfer resistance and consequently more efficient carrier migration. This result provides further evidence that the constructed heterojunction effectively facilitates electron transfer and enhances charge separation, thereby boosting the photocatalytic performance[33].
The separation of photogenerated carriers was further probed by photoluminescence (PL) spectroscopy. As shown in Fig.8c, under 320 nm excitation, 15%CNAT displayed a pronounced quenching of the PL intensity relative to g-C3N4 and TiO2. This quenching effect originates from the heterojunction formed at the g-C3N4/TiO2 interface, which creates an efficient charge-transfer pathway, thereby suppressing electron-hole recombination[34]. The resultant enhancement in charge separation and migration enables more photoinduced charges to participate in surface redox reactions, ultimately boosting the photocatalytic performance. The consistent findings from PL, photocurrent density, and EIS measurements unequivocally demonstrate the enhanced charge separation and transport efficiency of 15%CNAT, attributable to the synergistic effects within the heterojunction.
To identify the key active species in the photocatalytic process, scavenging experiments were performed, and the results are shown in Fig.9a. In the control experiment without any quenching agent, TC was significantly degraded within 300 min. The degradation efficiency was, however, notably suppressed upon the introduction of specific scavengers for ·O2- [captured by p-benzoquinone (BQ)], ·OH [captured by tert-butanol (TBA)], electrons (e-, captured by AgNO3), and holes [h+, captured by disodium ethylenediaminetetraacetate (EDTA-2Na)]. The most pronounced inhibition was observed with BQ and AgNO3, followed by TBA, while EDTA-2Na also caused a discernible reduction. These results confirm that ·O2-, ·OH, e-, and h+ all play crucial roles in the degradation process, with ·O2-, ·OH, and e- being the most dominant species.
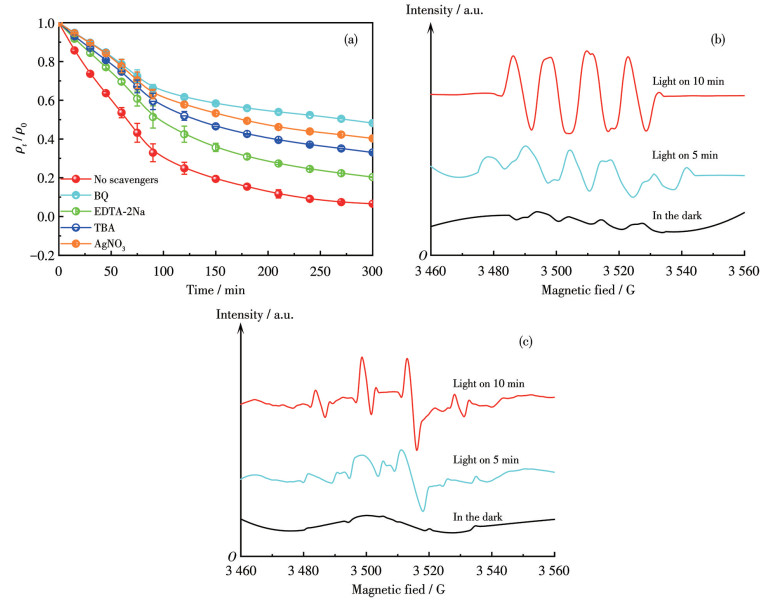
Electron paramagnetic resonance (EPR) measurements using DMPO as a spin-trapping agent were conducted to further verify the generation of reactive radicals. As depicted in Fig.9b, no characteristic signals for the DMPO-·O2- adduct were observed in the dark. Upon light irradiation for 5 min, a weak signal emerged, which intensified significantly after 10 min, confirming that ·O2- formation is photo-induced and accumulates in a time-dependent manner. Similarly, for ·OH (Fig.9c), distinct DMPO-·OH adduct signals were absent in the dark but became clearly visible after 5 min of illumination and strengthened after 10 min, demonstrating that ·OH generation was also light-driven and time-accumulative. In summary, the combined evidence from scavenging experiments and EPR analyses confirms that ·O2- and ·OH serve as the dominant active species. Their light-driven generation and time-dependent accumulation provide direct mechanistic insight into the photocatalytic process.
The conduction band (CB) energies (ECB) of g-C3N4 and TiO2 in 15%CNAT were calculated using the equation ECB=χ-Ee-0.5Eg[35], where χ is the geometric mean of the constituent atoms′ electronegativity, Ee is the energy of the standard hydrogen electrode (4.5 eV), and Eg is the band gap. The resulting ECB values were -0.505 eV for g-C3N4 and -0.195 eV for TiO2. The corresponding valence band (VB) energies (EVB) were then determined from EVB=ECB+Eg[36], yielding values of 1.805 eV for g-C3N4 and 2.915 eV for TiO2. According to Wang et al.[37], the work functions of TiO2 and g-C3N4 are 6.58 and 4.18 eV, respectively. Since the work function correlates directly with the Fermi level (EF), the Fermi level of TiO2 is initially lower than that of g-C3N4 before contact (Fig.10a). Upon heterojunction formation, electrons migrate spontaneously from g-C3N4 to TiO2 until their Fermi levels equilibrate (Fig.10b). This electron transfer depletes g-C3N4 of electrons, inducing a positive surface charge and upward band bending, while the electron accumulation in TiO2 causes a negative surface charge and downward band bending. Consequently, a built-in electric field is established across the g-C3N4/TiO2 interface. Under simulated sunlight irradiation, both g-C3N4 and TiO2 absorb photons, promoting electrons from the VB to the CB. Driven collectively by the built-in electric field, band bending, and Coulomb interaction, electrons in the CB of TiO2 tend to migrate and recombine with holes in the VB of g-C3N4. In contrast, the recombination between electrons in the CB of g-C3N4 and holes in the VB of TiO2 is effectively suppressed.
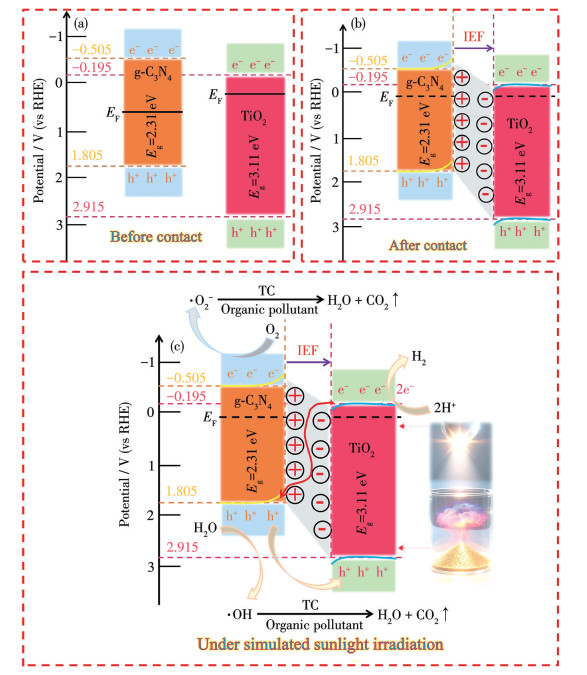
Fig. 10c schematically illustrates the S-scheme charge transfer mechanism within the g-C3N4/TiO2 heterojunction, showing the internal electric field (IEF)-driven charge transfer and separation. This mechanism selectively quenches the less reactive charge carriers—electrons in the TiO2 CB and holes in the g-C3N4 VB—thereby preserving the highly reactive electrons in the g-C3N4 CB and holes in the TiO2 VB. The resulting directional electron flow substantially enhances the overall photocatalytic redox efficiency, a conclusion supported by scavenger experiments and EPR analysis. Further corroboration is provided by the binding energy shifts detected in both in-situ and ex-situ XPS spectra (Fig. 2), which affirm the proposed charge migration pathway. Through the S-scheme heterojunction, the 15%CNAT catalyst surface is enriched with highly oxidative holes (h+). These h+ directly oxidize adsorbed TC or generate ·OH from H2O/OH-, thereby synergistically degrading TC functional groups (e.g., amide bonds, benzene rings) into small molecules and ultimately mineralizing them into CO2, H2O, and inorganic ions. Concurrently, CB electrons reduce O2 to ·O2-, which further transforms and participates in oxidative degradation, accelerating the process. Moreover, these accumulated electrons exhibit sufficient reducing power [with an energy below 0 eV (vs H+/H2)] to reduce H+ for hydrogen evolution. The visible-light response of g-C3N4 extends the spectral absorption range of the composite, enabling the utilization of visible light to generate more electron-hole pairs. The S-scheme heterojunction mechanism then efficiently separates these photogenerated charges, ensuring a sustained supply of electrons for H+ reduction while effectively suppressing efficiency losses from carrier recombination. Consequently, the S-scheme mechanism provides a more appropriate explanation for the enhanced photocatalytic activity observed in the xCNAT composites.
This study successfully constructed a series of S-scheme g-C3N4/TiO2 heterojunction photocatalysts using a combined thermal polymerization-hydrothermal-calcination method. The g-C3N4/TiO2 heterojunctions demonstrated markedly enhanced performance in both TC degradation and photocatalytic hydrogen evolution compared to TiO2 or g-C3N4 alone. This improvement is attributed to the synergistic effect of a porous structure, which provides abundant reactive sites, and efficient S-scheme charge separation. Mechanistic studies reveal that the S-scheme heterojunction enables the directional utilization of photogenerated carriers: driven by the built-in electric field, band bending, and Coulomb interactions, electrons accumulate in the TiO2 conduction band for H2 evolution, while holes are retained in the g-C3N4 valence band for TC oxidation. This work provides a feasible strategy for designing dual-functional photocatalysts that integrate environmental remediation with energy production, demonstrating the promising potential of the g-C3N4/TiO2 heterojunction for antibiotic wastewater treatment and clean energy generation. Future work may focus on optimizing scalable synthesis and assessing performance under real-world conditions.
Supporting information is available at
ZHAO M, LI W J, YANG M W, ZHAO Z H, YE R G, MAO X W, PADGETT P, CHEN P. Long-range enhancement of micropollutant adsorption on metal-promoted photocatalysts[J]. Nat. Catal., 2024, 7(8): 912-920 doi: 10.1038/s41929-024-01199-0
KLEIN E Y, IMPALLI I, POLEON S, DENOEL P, CIPRIANO M, VAN BOECKEL T P, PECETTA S, BLOOM D E, NANDI A. Global trends in antibiotic consumption during 2016—2023 and future projections through 2030[J]. Proc. Nat. Acad. Sci., 2024, 121(49): 1-12
HISATOMI T, YAMADA T, NISHIYAMA H, TAKATA T, DOMEN K. Materials and systems for large-scale photocatalytic water splitting[J]. Nat. Rev. Mater., 2025, 10(10): 769-782 doi: 10.1038/s41578-025-00823-0
XU Z S, ZHANG H B, ZHOU J Y, HUANG T, SUN J, CHANG C, CHEN Z, LIU K, TONG Z F. Sunlight-driven peroxydisulfate activation by floating Ca/Fe-TiO2@polystyrene spheres for efficient moxifloxacin degradation in circulated water: Critical roles of Ca/Fe bimetallic ions crosslinking on the stability of catalyst structure and electron transfer of system[J]. Chem. Eng. J., 2025, 513: 162750 doi: 10.1016/j.cej.2025.162750
WANG X S, YUAN Z Y. Insights into semiconductor heterojunctions for enhanced photocatalytic performance and applications[J]. Coordin. Chem. Rev., 2026, 548(Part 1): 217184
YANG P H, YANG J Q, ZEN X Y, GONG Y Y, ZHONG J B. Construction of S-scheme Sb2O3/g-C3N4 heterojunctions for photocatalytic CO2 and Cr(Ⅵ) reduction[J]. J. Colloid Interface Sci., 2025, 700(Part 3): 138648
SHAWKY A, MOHAMED R M. S-scheme heterojunctions: Emerging designed photocatalysts toward green energy and environmental remediation redox reactions[J]. J. Environ. Chem. Eng., 2022, 10(5): 108249 doi: 10.1016/j.jece.2022.108249
SONG C H, ZHANG Y T, ZHANG Z F, WANG H T, ZOU J. Synergistic carbonyl/vacancy engineering of aspartic acid-modified g-C3N4 nanorods for exceptional photocatalytic H2 production[J]. Chem. Eng. J., 2025, 525: 170465 doi: 10.1016/j.cej.2025.170465
GU S M, LI Y F, LIU Z P. Optimal interface structure in anatase-rutile mixed TiO2 for enhanced charge separation and prolonged carrier lifetime[J]. J. Chem. Phys., 2025, 163(21): 214701 doi: 10.1063/5.0299972
YANG Y Q, MA G S, AN Z J, WANG W, HU X L, WANG Y, DU Z L, GONG X Z, TAN H Y, GUO F X, TANG J G. Preparation of recyclable g-C3N4/TiO2 heterojunction/alginate hydrogelmicrobeads and investigation of their adsorption-photocatalytic properties[J]. J. Hazard. Mater. Adv., 2025, 18: 100650
QU W B, LOUANGSOUPHOM B, YE X L, LIU H M, WANG X. Rare earth/N co-dopants embedded g-C3N4/TiO2-macroporous resin composites for efficient adsorption-photocatalytic removal of microcystin-LR[J]. J. Environ. Chem. Eng., 2025, 13(5): 118632 doi: 10.1016/j.jece.2025.118632
HU C, HAO Y K, CHANG Y T, KAO L H, CHUANG K S, HUANG J H, WIBOWO A. Type-Ⅱ heterojunction of g-C3N4/TiO2 hollow spheres for photocatalytic H2 production and degradation of organic contaminants in water[J]. J. Alloy. Compd., 2025, 1032: 181104
GAN W, CHEN R X, ZHANG L, GUO J, ZHANG M, LU Y Q, SUN Z Q. Construction of S-scheme cyano-modified g-C3N4/TiO2 film with boosted charge transfer and highly hydrophilic surface for enhanced photocatalytic degradation of norfloxacin[J]. J. Mater. Sci. Technol., 2025, 206: 74-87 doi: 10.1016/j.jmst.2024.03.039
YANG Y Y, LIU X F, ZHOU Q, GAO T Y, WU X H, WANG G H. Dual roles of MHCNF in microwave-antenna induced synthesis and efficient photogenerated carriers transfer of g-C3N4/TiO2 heterojunction photocatalyst[J]. Appl. Surf. Sci., 2025, 712: 164186 doi: 10.1016/j.apsusc.2025.164186
LIU S R, YE Q J, LI Q, MA Z Z, YANG R, LI D, JIANG D L. Enhancing interfacial charge transfer efficiency via π-π interaction in PTA/VC-CN S-scheme heterojunction for efficient photocatalytic CO2 conversion[J]. Chem. Eng. J., 2025, 519: 164987 doi: 10.1016/j.cej.2025.164987
GUO Z X, QIN H Y, SHAN P N, XIONG B, WANG L J, SHI W L, SUN Y, GUO F. Photo-activated piezoelectric-catalyzed hydrogen peroxide production in pure water by carbon-modified graphitic carbon nitride[J]. Chem. Eng. J., 2025, 520: 165829 doi: 10.1016/j.cej.2025.165829
AI L S, ZHAO Z B, SONG X D, TANG Y C, FENG K, WANG X Z, ZHANG B L, BI H H, QIU J S. Curved-slit effect induced nucleation for g-C3N4 nanorings with double concaves and enhanced photocatalysis[J]. Adv. Funct. Mater., 2025, 35(37): 2506395 doi: 10.1002/adfm.202506395
ZOU Y D, YANG B B, LIU Y, REN Y, MA J H, ZHOU X R, CHENG X W, DENG Y H. Controllable interface-induced co-assembly toward highly ordered mesoporous Pt@TiO2/g-C3N4 heterojunctions with enhanced photocatalytic performance[J]. Adv. Funct. Mater., 2018, 50: 1806214
ZHOU Y M, YANG W L, FENG L J, HONG J, ABBAS M, KAWI S. Sunflower-disc-inspired vertical growth of 2D ZnIn2S4 on ultra-thin TiO2: Constructing a 3D porous photocatalytic glass film for ultra-efficient organic pollutant degradation[J]. Appl. Catal. B‒Environ., 2025, 363: 124782 doi: 10.1016/j.apcatb.2024.124782
XIN S S, MA X M, LU J R, ZHANG G S, HUO S Y, GAO M C, XU P, LIU W J, FU W X. Enhanced visible light photoelectrocatalytic degradation of o-chloronitrobenzene through surface plasmonic Au nanoparticles and g-C3N4 co-modified TiO2 nanotube arrays photoanode[J]. Appl. Catal. B‒Environ., 2023, 323: 122174 doi: 10.1016/j.apcatb.2022.122174
YOOM M, PARK Y, SIM H, KWON H, LEE Y, JANG H, CHOI S, SON J. 2D vacancy confinement in anatase TiO2 for enhanced photocatalytic activities[J]. Adv. Mater., 2025, 37(15): 2413062 doi: 10.1002/adma.202413062
YANG D X, XIA Y W, XIAO T, XU Z P, LEI Y F, JIAO Y, ZHU X D, FENG W. Constructing Ag-TiO2-g-C3N4 S-scheme heterojunctions for photocatalytic degradation of malachite green[J]. Opt. Mater., 2025, 159: 116652 doi: 10.1016/j.optmat.2025.116652
ABDALLAH S S, MARIDEVARU M C, MARZOUQI F, SELVARJ R. Green synthesis of TiO2@g-C3N4 nanocomposites for the photocatalytic degradation of pesticides and toxic organics present in water and wastewater[J]. J. Photochem. Photobiol. A‒Chem., 2025, 459: 116015 doi: 10.1016/j.jphotochem.2024.116015
SANTOS G, TIAN L, GONCALVES R, GARCIA H, ROSSI L. Boosting CO2 photoreduction efficiency of carbon nitride via S-scheme g-C3N4/Fe2TiO5 heterojunction[J]. Adv. Funct. Mater., 2025, 35(29): 2422055 doi: 10.1002/adfm.202422055
WU Q Q, LV D D, HU T Y, LI L, ZHANG W J, WANG H X, GAO Q Y, ZHAO Z N. Enhanced photocatalytic performance of gully-like AgI/Cu-TiO2 composites with synergistic effects of phase junction and S-scheme heterojunction[J]. J. Phys. Chem. Solids, 2026, 208(Part 2): 113197
ERDEM N G, CAGLAR B, LNAB E, TUNA Ö, ERTIS İ F, SIMSEK E B. Incorporation of CuWO4 with hollow tubular g-C3N4: Harnessing the potential in photocatalytic degradation, hydrogen production, and supercapacitor applications[J]. Renew. Energy, 2026, 256(Part D): 124235
HAZARAIMI M H, GOH P S, WANG L Y, LAU W J, SUBRAMANIAM M, NAIDUD M, ISMAIL A F, HASHIM N, KERISNAN N D, YAHAYA N M, MAMAT R R. Multifunctional roles of g-C3N4 in synthesizing N-TiO2/g-C3N4 heterojunction photocatalyst for photodegradation of bisphenol A[J]. Arab. J. Sci. Eng., 2025, 50(6): 1-15
ATACAN K, GUY N, SEMERCI A B. Designing green synthesis of silver nanoparticles from Citrus reticulata onto g-C3N4/TiO2 for antibacterial and photocatalytic activities[J]. Biocatal. Agric. Biotechnol., 2025, 64: 103524 doi: 10.1016/j.bcab.2025.103524
WANG M M, LI Q Y, LIU J G, GAO X, WEI Q, CHEN W J. Facile synthesis of novel 2D mesoporous g-C3N4/TiO2 heterojunction composites with high photocatalytic performance[J]. Ceram. Int., 2025, 51(20): 30948-30958 doi: 10.1016/j.ceramint.2025.04.287
HAN Y, LI Y J, LI T Y, ZHANG Q R, JIAO T F. Electrically enhanced adsorption and photocatalytic properties of g-C3N4/TiO2 heterostructure for removal of tetracycline in water: A study by molecular dynamics simulation and experiment[J]. J. Mol. Liq., 2024, 415(Part B): 126412
XUE X L, WANG C Y, ZHU X, YE D D, YANG Y, WANG H, CHEN R, LIAO Q. Visible-light responsive photocatalytic fuel cell with gas-diffusion g-C3N4/TiO2 heterojunction photoanode for simultaneously degrading VOCs and generating electricity[J]. J. Environ. Chem. Eng., 2025, 5: 114087
MUHAMMAD T. Well-designed V2AlC MAX supported g-C3N4/TiO2 Z-scheme heterojunction for photocatalytic CO2 reduction through bi-reforming to produce CO and CH4[J]. Energy, 2024, 310: 133231 doi: 10.1016/j.energy.2024.133231
XU Y F, LI Y, WEI Y J, LIANG H O, XU T, SUN Y H, BAI J. Facile synthesis of Pd loaded g-C3N4/TiO2 heterojunction catalyst with enhanced photothermal catalytic performance in Suzuki coupling under moderate condition[J]. Colloid. Surface A, 2024, 690: 133751 doi: 10.1016/j.colsurfa.2024.133751
LIANG S R, AN M Z, XIA S N, ZHANG B B, XUE B, XU G M. Enhanced photocatalytic degradation of methyl orange by TiO2/biochar composites under simulated sunlight irradiation[J]. Opt. Mater., 2023, 142: 114105 doi: 10.1016/j.optmat.2023.114105
JIANG X, JIANG H, TANG Y M, ZHANG H Z, YANG L B, WANG X W, ZHAO B. g-C3N4/TiO2-X heterojunction with high-efficiency carrier separation and multiple charge transfer paths for ultrasensitive SERS sensing[J]. Chin. Chem. Lett., 2024, 35(10): 109415 doi: 10.1016/j.cclet.2023.109415
MAITLO H A, YOUNIS S A, KIM K. Understanding oxidation potential and degradation mechanism of acid-treated TiO2 coupled g-C3N4 S-scheme heterojunction photocatalyst for the removal of gaseous formaldehyde[J]. Sep. Purif. Technol., 2025, 356(Part A): 129862
WANG J, WANG G H, CHENG B, YU J G, FAN J J. Sulfur-doped g-C3N4/TiO2 S-scheme heterojunction photocatalyst for Congo red photodegradation[J]. Chin. J. Catal., 2021, 42(1): 56-68 doi: 10.1016/S1872-2067(20)63634-8

Figure 1 (a) XRD patterns, (b) nitrogen adsorption-desorption isotherms, (c) corresponding pore-size distribution curves, and (d) FTIR spectra of g-C3N4, TiO2, and xCNAT

Figure 2 In-situ and ex-situ XPS spectra of g-C3N4, TiO2, and 15%CNAT: (a) survey, (b) C1s, (c) N1s, (d) Ti2p, and (e) O1s

Figure 3 Electron microscopy characterization of the synthesized materials: SEM images of (a) g-C3N4, (b) TiO2, (c, d) 15%CNAT; (e, f) TEM images, (g) HRTEM image, and (h) corresponding SAED pattern of 15%CNAT

Figure 4 (a, b) SEM images and (c-f) EDS elemental mappings, and (g) EDS spectrum for 15%CNAT

Figure 6 (a) Performance photocatalytic degradation of TC over different photocatalysts; (b) Effect of 15%CNAT dosage on the photocatalytic degradation of TC; (c) Effect of solution pH on the photocatalytic degradation of TC; (d) Effect of initial TC mass concentration on the photocatalytic degradation of TC

Figure 7 (a) Hydrogen evolution under simulated sunlight irradiation as a function of catalyst dosage (reaction conditions: t=8 h, 5 mL TEOA, 100 mL H2O, 300 W Xe lamp); (b) Cycling measurements of 15%CNAT under simulated sunlight irradiation (reaction conditions: t=8 h, 100 mg catalyst, 5 mL TEOA, 100 mL H2O, 300 W Xe lamp)

Figure 8 (a) Transient photocurrent responses of the samples under xenon lamp illumination; (b) EIS Nyquist plots and (c) PL spectra of the as-prepared samples measured under 320 nm excitation

Figure 9 (a) Photocatalytic activities of 15%CNAT for TC degradation with disparate scavengers; EPR spectra of 15%CNAT for (b) DMPO-·O2- and (c) DMPO-·OH

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

