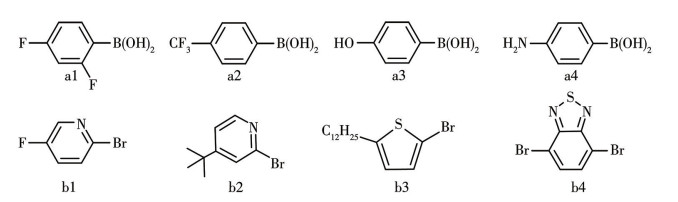
图 1.
C—C偶联反应的底物
Figure 1.
Substrates of the C—C coupling reaction

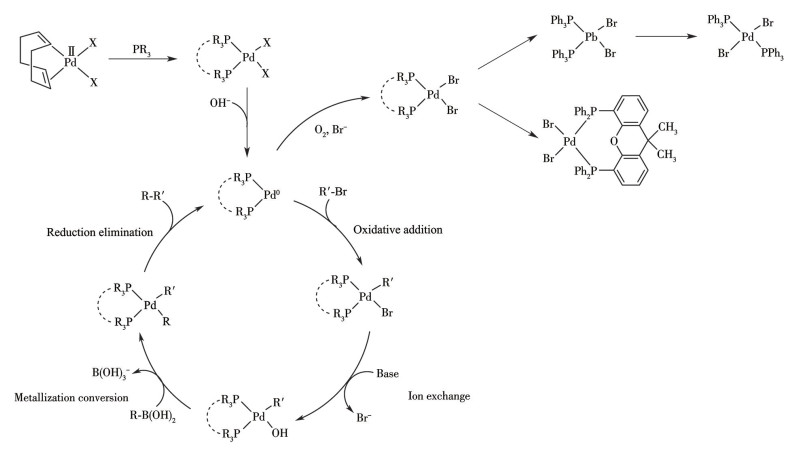
基于大位阻富电子的膦和NHC卡宾等配体调控的Pd(Ⅱ)催化前驱体还原新生Ln-Pd(0)催化剂,是实现钯催化剂高效活化的关键步骤[1-3],也是提升催化反应中底物转化率与选择性等的核心策略[4-5]。已有大量研究以PdCl2、Pd(OAc)2、Pd(PPh3)4和Pd1-2(dba)2-3等基础钯化合物或对它们进行简单修饰得到的Pd(L)X2(L=配体,X=卤素)为前驱体,通过考察氮配体(如季铵盐[6]、双吡啶胺类[7])或膦配体[8-9](如PPh3、P(t-Bu)3或手性膦)的辅助作用、底物选择以及生成的钯过渡态等因素,探究对偶联反应底物的转化率或选择性。Wang等[10]通过引入膦配体,克服了Pd(L)X2等前驱体因新生Pd(0)的团聚为钯黑失活等缺点,在对映选择性三组分反应中实现高收率和高选择性。近期研究发现,在膦配体参与下,Pd(Ⅱ)前驱体还原过程中形成的卤素桥联Pd(Ⅰ)有机膦二聚体中间体,被认为是偶联反应的最有效催化剂[11]。此外,已有大量报道证明具有大位阻的富电子配体是提高偶联反应的活性,实现高对映选择性的关键手段[12],这归因于富电子配体的电子效应和空间位阻效应对Pd中心的调控[13]。配体和卤素是Pd(Ⅱ)前驱体的重要组成部分,关于其如何协同前驱体实现新生Ln-Pd(0)可控新生和新生特定结构的Ln-Pd(0)作用于C—C偶联反应的研究虽然已有部分报道[14-15],但缺乏深入研究,更缺乏卤素协同下,PR3配体调控的COD-Pd前驱体(COD=1,5-环辛二烯)对C—C偶联反应的作用机制研究。
本研究以[Pd(COD)X2](X=Cl、Br)为催化前驱体,分别在单膦(PPh3)和双膦(Xantphos)配体的调控下,考察其在C—C偶联反应中的催化性能。综合运用元素分析、红外光谱、核磁共振氢谱、密度泛函理论(DFT)计算和紫外可见光谱,研究[Pd(COD)X2]分子中Pd(Ⅱ)中心的化学结构及其在常见溶剂中的稳定性;通过单晶X射线衍射解析反应后催化剂中Pd(Ⅱ)中心的配位构型,并结合[Pd(COD)X2]的晶体结构[16]和Pd(Ⅱ)催化C—C偶联反应的机理,重点探究PR3和COD的结构及卤素原子对原位新生Ln-Pd(0)的调控作用,揭示前驱体Pd(Ⅱ)中心的化学结构在催化循环中的演变及关键作用机制。
所用的主要仪器有Avance Ⅲ核磁共振仪(500MHz,TMS为内标,瑞士Bruker公司)、Apex Duo双光源单晶衍射仪(德国布鲁克AXS公司)、Bruker Tensor-27型傅里叶变换红外光谱仪(KBr压片,400~4 000 cm-1)、Varian Carry50型紫外可见分光光度计、Agilent 5110电感耦合等离子体发射光谱仪(安捷伦科技有限公司)。卤化钯(PdX2,X=Cl、Br)购自贵金属集团,其它试剂均为市售分析纯。
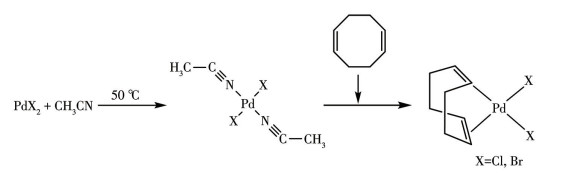
在500 mL的双颈反应瓶中,加入乙腈(CH3CN) 100 mL、10 mmol PdX2,50 ℃下搅拌直到PdX2完全溶解,加入46 mmol的COD,再搅拌30 min后停止加热并冷却到室温。采用布氏漏斗抽滤收集沉淀产物。粗产物用二氯甲烷(DCM)溶解后,经0.22 μm聚四氟乙烯微孔滤膜过滤除去不溶性杂质。滤液经旋转蒸发仪浓缩,得到粉末状固体,产物在45 ℃下真空干燥4 h,称重,基于钯计算产率。
[Pd(COD)Cl2]:黄色粉末,2.74 g,产率96%。元素分析按C8H12Cl2Pd的计算值(%):C,33.65;H,4.24;Cl,24.83;Pd,37.28。实测值(%):C,33.71;H,4.32;Cl,24.46;Pd,37.26。1H NMR(500 MHz,CDCl3):δ 6.32(s,4H),2.92(d,J=12.5 Hz,4H),2.57(d,J=9.7 Hz,4H)。13C NMR(125 MHz,CDCl3):δ 116.71,30.99。IR(cm-1):3 436[ν(O—H)];3 045[ν(=C—H)];3 013~ 2 883[ν(—C—H)]; 1 523~1 422[ν(C=C)]; 1 343[δ(CH2)]; 1 179,1 087[δ(C—H)];994[νs(ring)];791[ν(Pd—Cl)+ δ(ring)];453[ν(Pd—Cl)]。
[Pd(COD)Br2]:酒红色粉末,3.54 g,产率为94%。元素分析按C8H12Br2Pd的计算值(%):C,25.66;H,3.23;Br,42.68;Pd,28.43。实测值(%):C,25.71;H,3.27;Br, 42.62;Pd, 28.43。1H NMR(500 MHz,CDCl3):δ 6.39(s,4H),2.80(d,J=12.5 Hz,4H),2.46(s,4H)。13C NMR(125 MHz, CDCl3):δ 116.60,31.17。IR(cm-1):3 466[ν(O—H)];3 048[ν(=C—H)];3 011~2 882 [ν(—C—H)];1 525~1 420[ν(C=C)];1 343[δ(CH2)];1 176、1 086[δ(C—H)];991[νs(ring)];786[ν(Pd-Br)+ δ(ring)];447[ν(Pd-Br)]。
以市售trans-[Pd(PPh3)2Cl2]为参照,PR3(PR3=PPh3、Xantphos)为膦配体,[Pd(COD)X2]为前驱体,溴代芳基和苯硼酸衍生物为底物(图 1),在相同的实验条件下平行实验[17]。
将[Pd(COD)X2]前驱体(x=1%)、PR3(1.0倍~2.0倍物质的量)、干燥的四氢呋喃(0.1 mol·L-1)依次加入50 mL干燥的双颈圆底烧瓶中。混合物在室温下搅拌10 min,完成Pd前驱体的活化。将碳酸钠(2.0倍物质的量)溶于水形成溶液(0.4 mol·L-1),随后向上述混合物中依次加入苯硼酸衍生物(1.0倍物质的量)、2-溴吡啶衍生物(0.8倍物质的量)和碳酸钠水溶液,氮气交换3次,置于90 ℃下搅拌反应3~5 h。反应体系冷却至室温,旋蒸去除大量溶剂。水相用乙醚(100 mL)萃取3次,合并有机萃取液,用饱和盐水(500 mL)洗涤,经无水硫酸钠干燥后减压浓缩。粗产物通过硅胶柱层析法进行纯化(洗脱剂:石油醚(PE)/乙酸乙酯(EA),体积比10∶1),得到目标产物。最后使用纯DCM作为洗脱剂回收得到催化剂的DCM溶液,旋蒸得到固体催化剂,整个过程以TLC检测。
分别对前驱体[Pd(COD)X2]和催化反应后回收的催化剂采用溶剂缓慢挥发法培养其单晶。选取大小为0.20mm×0.17mm×0.16 mm([Pd(COD)Cl2])、0.16 mm×0.11 mm×0.06 mm([Pd(COD)Br2])、0.20 mm×0.17 mm×0.16 mm(trans-[Pd(PPh3)2Br2])和0.16 mm×0.11 mm×0.06 mm(cis-[Pd(Xantphos)Br2])的晶体,在Bruker APEX-Ⅱ CCD型单晶X射线衍射仪上,采用经石墨单色器化的Cu Kα射线(λ=0.154 178 nm),于150(2) K,以θ-ω扫描方式在设定的角度范围内收集衍射数据。衍射数据用SADABS程序进行经验吸收校正,将所有前驱体的单晶衍射数据在OLex2上用SHELXT-2018程序解析,对所有前驱体的非氢原子坐标及其各向异性温度因子用SHELXL程序进行全矩阵最小二乘法修正至收敛,所有氢原子均为理论加氢。
Benchekroun等[18]早于1977年便解析了[Pd(COD)Cl2]和[Pd(COD)Br2]的晶体结构,确认为顺式配位构型。出于本工作对比研究的需要,重新合成了[Pd(COD)Cl2]和[Pd(COD)Br2],并培养了适用于单晶X射线衍射分析的单晶,其晶体学数据列于Supporting information。trans-[Pd(PPh3)2Br2]和cis-[Pd(Xantphos)Br2]的主要晶体学数据列于表 1。
 下载:
导出CSV
下载:
导出CSV
| Parameter | trans-[Pd(PPh3)2Br2] | cis-[Pd(Xantphos)Br2] |
| Empirical formula | C36H30Br2P2Pd·2CH2Cl2 | C39H32Br2OP2Pd·C4H8O2 |
| Formula weight | 960.61 | 932.91 |
| Crystal system | Orthorhombic | Triclinic |
| Space group | Pbca | P1 |
| a / nm | 2.017 58(9) | 1.032 33(4) |
| b / nm | 0.807 92(3) | 1.143 37(5) |
| c / nm | 2.322 16(9) | 1.722 37(7) |
| α / (°) | 79.611(2) | |
| β / (°) | 87.514(2) | |
| γ / (°) | 83.738(2) | |
| Volume / nm3 | 3.785 2(3) | 1.987 13(14) |
| Z | 4 | 2 |
| Dc / (Mg·m-3) | 1.686 | 1.559 |
| μ / mm-1 | 10.035 | 7.183 |
| F(000) | 1 904 | 936 |
| θ range / (°) | 4.38-68.95 | 2.61-68.66 |
| Index ranges | -24 ≤ h ≤ 22, -9 ≤ k ≤ 9, -27 ≤ l ≤ 27 | -12 ≤ h ≤ 12, -13 ≤ k ≤ 13, -17 ≤ l ≤ 20 |
| Reflection collected | 23 098 | 28 080 |
| Independent reflection | 3 480 (Rint=0.107 0) | 7 268 (Rint=0.076 8) |
| Goodness-of-fit on F 2 | 1.078 | 1.070 |
| Final R indices [I > 2σ(I)] | R1=0.035 4, wR2=0.085 5 | R1=0.052 4, wR2=0.151 8 |
| R indices (all data) | R1=0.052 7, wR2=0.090 7 | R1=0.066 2, wR2=0.166 0 |
| Largest diff. peak and hole / (e·nm-3) | 531 and -569 | 1 088 and -1 052 |
现有的[Pd(COD)X2]合成方法多采用高沸点溶剂,产率较低,钯回收利用率低。本研究所选的CH3CN兼具溶剂与配体的双重功能,PdX2的溶解过程是CH3CN与Pd(Ⅱ)配位生成trans-[Pd(CH3CN)2X2],CH3CN与Pd(Ⅱ)的配位作用较弱,双齿配体COD迅速取代CH3CN,通过2个C=C双键的π电子与Pd(Ⅱ)中心配位形成螯合环,并促使反式结构翻转为顺式,得到前驱体cis-[Pd(COD)X2](图 2)。通过元素分析、IR和1H NMR对它们的结构进行表征,证实所得产物为目标前驱体。
在前驱体的谱学表征中,卤素原子电负性和原子半径差异使它们的IR和1H NMR谱图表现规律性变化[19]。IR光谱中,Pd—Cl振动吸收为453 cm-1,而ν(Pd—Br)则红移至447 cm-1。1H NMR谱在δ=2.5~2.9的多组峰为COD的8个亚甲基(—CH2—)质子,δ=6.3的单峰为4个烯烃(=CH—)质子。与[Pd(COD)Cl2]相比,[Pd(COD)Br2]的1H NMR信号均向高场移动,这是由于溴原子相较于氯原子是更软、电负性更弱的配体,更强的给电子效应提高了Pd(Ⅱ)中心的电子云密度,增强了Pd中心向COD配体反键π轨道的反馈作用,使Pd—Br的IR吸收红移,也提高了烯烃氢的电子云密度,产生更强的屏蔽效应。
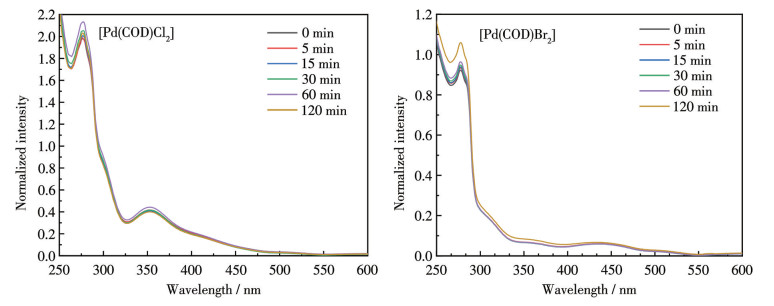
在室温下,2种Pd(Ⅱ)前驱体在THF及DCM溶剂中长时间放置后,UV-Vis光谱未发生明显变化,表现出较好的溶剂稳定性(图 3和4)。卤原子的孤对电子向Pd(Ⅱ)空d轨道跃迁产生的单重态金属-配体电荷转移(1MLCT)在350~500 nm有强吸收峰,300 nm处的强吸收则源于COD内部的π→π*跃迁[20]。与[Pd(COD)Cl2]相比,[Pd(COD)Br2]的特征吸收峰均发生约50 nm的红移。Br的4p轨道能级高于Cl的3p轨道且电离能更低,其孤对电子更易激发至Pd(Ⅱ)中心,促进了配体-金属电荷转移(LMCT)。且Br的原子半径更大,电子云更分散,与Pd(Ⅱ)的电子离域程度更强,LMCT跃迁的能垒降低,这表明Br能为Pd(Ⅱ)中心提供更高的电子云密度,更容易使Pd(Ⅱ)还原新生为Pd(0)。
采用ORCA量子化学软件,围绕Pd(Ⅱ)中心的化学结构特征对2种前驱体进行理论计算,研究COD和Pd—X键对Pd(Ⅱ)中心的影响及差异。以Neese等[21-23]的工作为参考,以泛函选择及结构解析为基础,对[Pd(COD)X2]进行几何优化并分析分子的轨道分布。计算结果表明:优化结构后[Pd(COD)X2]的Pd—X键长(0.229 2~0.243 2 nm,表 2)与已报道的晶体结构实验值(0.230 3~2.424 3 nm)[24]无偏差,表明该方法能准确呈现[Pd(COD)X2]的基态几何构型,模拟Pd—X键中卤原子差异对Pd(Ⅱ)中心化学结构和电子云密度影响。
下载:
导出CSV
| Precursor | Bond | Bond length / nm | |
| Experimental | Theoretical | ||
| [Pd(COD)Cl2] | Pd—Cl1 | 0.230 3 | 0.231 5 |
| Pd—Cl2 | 0.230 9 | 0.229 2 | |
| [Pd(COD)Br2] | Pd—Br1 | 0.242 5 | 0.241 8 |
| Pd—Br2 | 0.244 3 | 0.243 2 | |
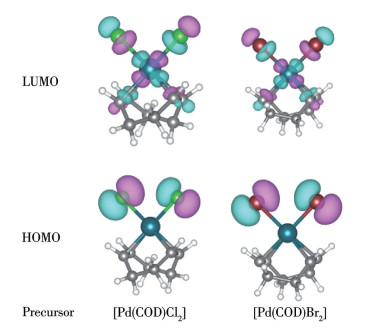
通过量子化学计算对[Pd(COD)X2]的电子结构特征分析表明(图 5):2种Pd(Ⅱ)前驱体的HOMO主要分布在Pd(Ⅱ)中心与相邻的卤素原子周围,与COD配体的部分区域有弱相互作用;Br原子半径比Cl大且电负性比Cl小,两者轨道的空间范围和密度略有差异。进一步研究发现,HOMO大部分定位于Cl的3p和Br的4p轨道,LUMO以卤素反键轨道和Pd(Ⅱ)的d轨道为主,COD的π轨道几乎不参与。由表 3可得:[Pd(COD)Cl2]的HOMO能级为-6.844 eV,LUMO能级为-2.852 eV,ΔE为3.992 eV,[Pd(COD)Br2]的HOMO能级则升高0.501 eV至-6.343 eV,能级差异表现显著,表明Br配体增强了Pd(Ⅱ)中心电子密度,LUMO能级降低0.098 eV至-2.950 eV,同时能隙缩小0.600 eV至3.392 eV,直接导致紫外吸收光谱发生50 nm红移,其中HOMO升高贡献率达83%。
下载:
导出CSV
| Precursor | E / eV | Bandgap (ΔE) / eV | |
| HOMO | LUMO | ||
| [Pd(COD)Cl2] | -6.844 | -2.852 | 3.992 |
| [Pd(COD)Br2] | -6.343 | -2.950 | 3.392 |
在前驱体[Pd(COD)X2]中,COD作为η2-π配体,通过其C=C双键的π电子云与Pd中心配位,是σ给电子与π反馈键共同作用;Pd—X则以σ配位键为主,伴随弱π反馈。大位阻富电子膦配体PR3主要通过强σ给电子作用与Pd中心形成Pd—P σ配位键,其空σ或π*轨道可接受Pd中心的反馈电子形成弱π键。DFT计算表明,[Pd(COD)X2]的HOMO主要来自X的σ给电子轨道,LUMO主要来自X或Pd的d轨道,COD与Pd中心的键合以弱动态相互作用为主;而Pd—X键因X的轨道与分子的HOMO紧密重叠,两者电子云强烈融合,形成了牢固的σ键,键合更稳定。因此,当PR3配体进攻Pd(Ⅱ)中心,COD因与Pd的前线轨道重叠弱、键合松弛,比X更易被取代,最终形成新的Pd—PR3 σ配位键。
以市售的Pd(PPh3)2Cl2为参照物(反式结构更稳定,市售多为trans-[Pd(PPh3)2Cl2]),PPh3和Xantphos为膦配体[25-26],考察[Pd(COD)X2]前驱体在Suzuki偶联反应中的催化活性(表 4~8)。
下载:
导出CSV
 |
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 79 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 10 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 1.5 | 82 |
| 4 | [Pd(COD)Cl2] | Xantphos | 1.5 | 94 |
| 5 | [Pd(COD)Br2] | 12.0 | 15 | |
| 6 | [Pd(COD)Br2] | PPh3 | 1.5 | 88 |
| 7 | [Pd(COD)Br2] | Xantphos | 1.5 | 97 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载:
导出CSV
 |
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 61 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 10 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 5.0 | 83 |
| 4 | [Pd(COD)Cl2] | Xantphos | 3.0 | 95 |
| 5 | [Pd(COD)Br2] | 12.0 | 15 | |
| 6 | [Pd(COD)Br2] | PPh3 | 5.0 | 84 |
| 7 | [Pd(COD)Br2] | Xantphos | 3.0 | 97 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载:
导出CSV
 |
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 73 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 16 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 5.0 | 80 |
| 4 | [Pd(COD)Cl2] | Xantphos | 3.0 | 92 |
| 5 | [Pd(COD)Br2] | 12.0 | 20 | |
| 6 | [Pd(COD)Br2] | PPh3 | 5.0 | 83 |
| 7 | [Pd(COD)Br2] | Xantphos | 3.0 | 92 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载:
导出CSV
 |
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 77 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 15 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 5.0 | 79 |
| 4 | [Pd(COD)Cl2] | Xantphos | 3.0 | 94 |
| 5 | [Pd(COD)Br2] | 12.0 | 23 | |
| 6 | [Pd(COD)Br2] | PPh3 | 5.0 | 81 |
| 7 | [Pd(COD)Br2] | Xantphos | 3.0 | 96 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载:
导出CSV
 |
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 80 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 21 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 5.0 | 83 |
| 4 | [Pd(COD)Br2] | 12.0 | 32 | |
| 5 | [Pd(COD)Br2] | PPh3 | 5.0 | 89 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
结果表明,在PR3配体参与下,以[Pd(COD)X2] (X=Cl、Br)为前驱体催化Suzuki反应,其产物收率显著高于无配体参与时的条件,优于市售催化剂trans-[Pd(PPh3)2Cl2]。其中,双齿配体Xantphos的效果优于单齿PPh3,且以[Pd(COD)Br2]为前驱体时收率优于[Pd(COD)Cl2]。
进一步分析表明,无配体参与条件下反应12 h后产物收率显著偏低,而引入PR3后反应效率显著提升,产率稳定达到80%以上。这表明富电子的PR3与Pd(0)的空d轨道形成强配位键,PR3通过空间位阻效应防止新生Pd(0)聚集为钯黑而失活。无膦配体添加时,新生的Pd(0)具有高表面活性,易快速团聚形成钯黑,该过程具有不可逆性,导致反应时间延长,无法提高产物的收率。商业催化剂trans-[Pd(PPh3)2Cl2]因本身具备稳定配位结构,表现出稳定的催化活性。
不同膦配体对钯催化偶联反应性能的影响规律结果表明,双齿膦配体Xantphos的催化表现最优,其对应的[Pd(COD)X2]/Xantphos体系产物收率高于[Pd(COD)X2]/PPh3体系,且反应时间更短;而[Pd(COD)X2]/PPh3体系的收率虽优于市售trans-[Pd(PPh3)2Cl2],但差异未达显著水平。这主要归结于刚性骨架的双膦配体Xantphos取代并延续COD与Pd的顺式配位结构,被还原新生为高活性的cis-(Xantphos)-Pd(0),其顺式结构更利于暴露更多的Pd(0)活性位点。单膦配体PPh3取代COD形成cis-[Pd(PPh3)2X2],该构型释放约25 kJ·mol-1的能量,易转化为trans-[Pd(PPh3)2X2],被还原新生的Pd(0)为cis-(PPh3)2-Pd(0)和trans-(PPh3)2-Pd(0)的混合物。市售的trans-[Pd(PPh3)2Cl2]则仅被还原为trans-(PPh3)2-Pd(0),该结构中2个PPh3将Pd(0)夹在中心,与cis-(PPh3)2-Pd(0)相比,暴露的Pd(0)活性位点降低,在偶联反应中cis-(PR3)2-Pd(0)比trans-(PR3)2-Pd(0)能获得更高的产物收率。
此外,在相同配体条件下,[Pd(COD)Br2]的收率普遍高于[Pd(COD)Cl2],这是由于Pd—Br键的断裂能低于Pd—Cl键,从而更易新生为Pd(0),表明催化剂的活性对卤素具有一定依赖性。
结合C—C偶联反应机理,解析反应前后催化剂的Pd中心价态和结构变化,是研究前驱体Pd(Ⅱ)中心的活化路径、Ln-Pd(0)形成机理及催化循环的关键。已确认[Pd(COD)Cl2]和[Pd(COD)Br2][18]的Pd(Ⅱ)中心为顺式构型,本文将直接引用。催化反应后回收的2种钯催化剂的主要键长和键角列于表 9,晶体结构如图 6和7所示。
下载:
导出CSV
| trans-[Pd(PPh3)2Br2] | |||||
| Pd—Br1 | 0.242 62 | Pd—P1 | 0.232 65 | P1—C1 | 0.181 34 |
| P1—C13 | 0.182 52 | ||||
| Br1—Pd1—P1 | 92.631 | Pd1—P1—C1 | 161.839 | C1—P1—C7 | 93.968 |
| C7—P1—C13 | 161.328 | ||||
| cis-[Pd(Xantphos)Br2] | |||||
| Pd—Br1 | 0.220 38 | Pd—Br2 | 0.222 57 | Pd—P1 | 0.222 39 |
| Pd—P2 | 0.217 69 | P1—C1 | 0.242 58 | P1—C8 | 0.244 30 |
| P1—Pd—Br2 | 85.867 | P2—Pd—Br1 | 84.934 | P1—Pd—P2 | 100.757 |
| Br2—Pd—Br1 | 86.521 | ||||
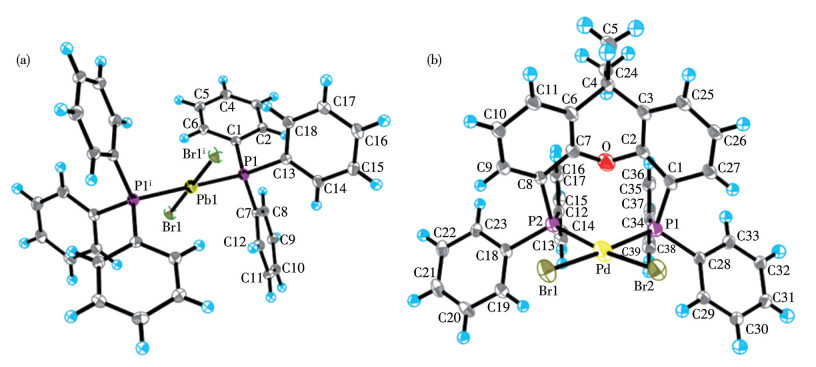
Symmetry code: ⅰ 1-x, 1-y, 1-z.
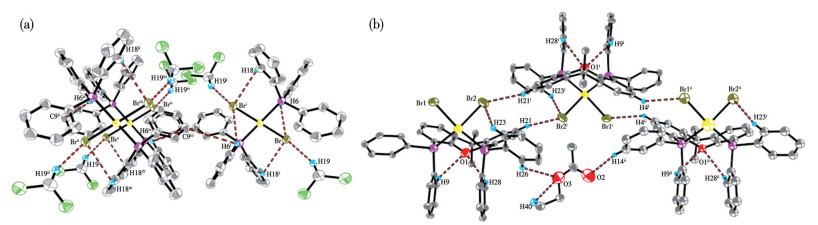
Symmetry codes: ⅰ 1-x, 1-y, 1-z; ⅱ 1/2-x, 1/2+y, z; ⅲ-1/2+x, 3/2-y, 1-z; ⅳ-1/2+x, 1/2-y, 1-z; ⅴ 1/2-x, -1/2+y, z for trans-[Pd(PPh3)2Br2]; ⅰ 1-x, 2-y, 1-z; ⅱ x, y, -1+z for cis-[Pd(Xantphos)Br2].
单晶X射线衍射分析表明,trans-[Pd(PPh3)Br2]属于正交晶系,空间群为Pbca;其配位几何构型为四方平面,中心Pd原子与2个PPh3配体及2个Br原子呈反式配位模式:2个PPh3配体分别占据四方平面的轴向位置,2个Br原子则处于赤道平面,形成对称的trans-[Pd(PPh3)Br2]配位单元。cis-[Pd(Xantphos)Br2]属于三斜晶系,空间群为P1;中心Pd原子通过与Xantphos配体的η2-双齿螯合形成四方平面配位构型。Xantphos配体的固有刚性使螯合环与Pd中心配位时发生轻微扭曲,降低分子的对称性。Xantphos配体的膦与氧桥的立体排斥和弯曲桥联结构使cis-[Pd(Xantphos)Br2]分子呈现显著的空间位阻和不规则构型,削弱了分子间作用力的方向性,促使晶体优先沿三斜晶系的低对称性空间群(P1)堆积。
trans-[Pd(PPh3)2Br2]中Pd—Br反式键长为0.242 62 nm,Pd—P反式键长为0.232 65 nm;[Pd(Xantphos)Br2]中Pd—Br键长(0.220 38和0.222 24 nm)与Pd—P键长(0.225 70和0.217 69 nm)均缩短。trans-[Pd(PPh3)2Br2]中,2个PPh3呈反式配位,通过反式效应削弱Br与Pd中心的键合,每个PPh3的强σ给电子作用竞争Pd的d轨道电子密度,反式Br的成键轨道电子云密度降低,Pd—Br键拉长。而cis-[Pd(Xantphos)Br2]中,Xantphos的2个P原子呈顺式配位,双齿螯合环的刚性结构减少了配体间的电子排斥,Pd—Br键长更短。PPh3与Pd的键合依赖Pd—P σ键,且因苯基的空间位阻,2个PPh3配体间存在弱反式排斥,进一步拉长Pd—P键。相比之下,Xantphos的双齿螯合结构通过“螯合环张力释放”形成热力学稳定配位球:2个P原子与Pd中心形成螯合环,成键效应增强Pd—P键强度,刚性骨架限制配体运动,减少空间拥挤导致的键长扩张,最终表现为更短的Pd—P键长。
在trans-[Pd(PPh3)2Br2]和cis-[Pd(Xantphos)Br2]的晶体中,分子堆积是多种弱相互作用力协同与竞争的结果。PR3配体主要通过范德瓦耳斯力和π相互作用(C—H…π)实现紧密堆积,而Br则通过卤键或弱的C—H…Br氢键参与构建特定的堆积模式[26-27]。如图 8所示,在trans-[Pd(PPh3)2Br2]的晶体堆积结构中,1个PPh3配体的其中一个苯环与相邻分子的C—H键通过C—H…π作用形成弱相互作用。该作用的几何参数如下:2个参与作用的环质心坐标分别为Cg1(0.157,0.596,0.503)和Cg2(-0.157,0.403,0.496),质心至氢原子的距离均为0.365 nm。相比之下,cis-[Pd(Xantphos)Br2]的晶体堆积中存在4种不同的C—H…π作用:膦配体上的2个苯环分别与相邻分子苯环上的氢原子形成此类作用。4个环质心的坐标分别为Cg1(0.915,0.770,0.520)、Cg2(0.084,1.229,0.479)、Cg3(0.153,1.199,-0.043)、Cg4(0.846,0.800,0.043),Cg1与H21(或Cg2与H21A)的距离为0.439 nm,Cg3与H4B(或Cg4与H4A)的距离为0.301 nm。上述晶体堆积结构的差异源于2种配合物中Pd(Ⅱ)中心的配位模式不同:PPh3作为单齿配体与Pd(Ⅱ)结合,形成动态但相对松散的配位环境;而Xantphos通过双齿螯合作用与Pd(Ⅱ)配位,构建了刚性且电子云密度更高的钯中心。当两者在反应中经原位还原生成(PR3)n-Pd(0)中间体时,相较于PPh3配体,Xantphos不仅能更有效地稳定Pd(0)物种,还可暴露更多活性位点。
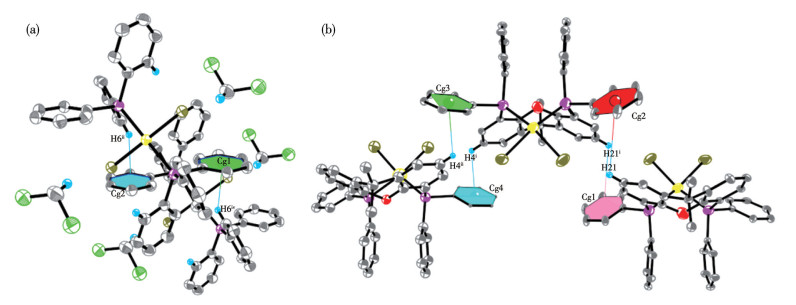
Symmetry codes: ⅱ 1/2-x, 1/2+y, z; ⅳ-1/2+x, 1/2-y, 1-z for trans-[Pd(PPh3)2Br2]; ⅰ 1-x, 2-y, 1-z; ⅱ x, y, -1+z for cis-[Pd(Xantphos)Br2].
基于上述的实验和理论计算结果,发现在反应过程中催化前驱体[Pd(COD)X2][28]的顺式构型被PR3配体取代并维持,形成cis-[Pd(PR3)2X2]中间体。在底物的作用下,cis-[Pd(PR3)2X2]和Pd—X通过协同作用实现顺式构型的Pd(0)新生,新生的cis-(PR3)2-Pd(0)与溴代底物C—X氧化加成得到cis-(PR3)2(R-)Pd(Ⅱ)-X中间体,再经过去质子化、转金属化、还原消除实现整个偶联反应的循环过程[29],cis-(PR3)2-Pd(0)实现了对底物的精准地抓取、组装并释放最终产物,如图 9所示。
催化前驱体[Pd(COD)X2]结构中的2个Pd-X(X=Cl、Br)维持催化前驱体在“沉睡”时的稳定性,被“唤醒”后作为牺牲配体实现Pd(Ⅱ)可控新生为Pd(0)。与Cl相比,Br的电负性小,对电子的束缚能力弱,能为Pd中心提供的电子云密度更大,为新生的Pd(0)提供更大的电子云密度,防止中心Pd(0)被氧化,维持高活性Pd(0)催化剂结构的相对稳定。此外,Pd—Br键更长,键更弱,更易解离,能提高催化前驱体活性位点的暴露效率[30]。
不添加辅助配体,[Pd(COD)X2]被新生为Pd(0)时,卤素和COD均作为牺牲配体离去,致使新生的Pd(0)极容易因团聚失活,导致偶联产物的收率较低[31]。添加大位阻富电子的PR3配体后,直接取代COD与Pd(Ⅱ)配位形成高能垒的cis-[Pd(PR3)2X2],而市售的trans-[Pd(PPh3)2X2]由PdX2与PPh3反应制备,该反式构型需克服约25 kJ·mol-1的能量才能转化为cis-[Pd(PPh3)2X2][32]。由于催化过程中存在cis-(PPh3)2-Pd(0)翻转为trans-(PPh3)2-Pd(0),导致[Pd(COD)X2]/PPh3较市售trans-[Pd(PPh3)2Cl2]对产物产率的提高不显著。相反,Xantphos的刚性骨架更能维持cis-(Xantphos)-Pd(0)的稳定性且提高了活性中心Pd(0)的暴露,获得更高的产物收率[33]。
在Pd-PR3片段中,富电子的PR3配体向钯中心提供更多电子密度,使新生的cis-(PR3)2-Pd(0)不易被氧化,PR3的大空间位阻物理性地保护新生的Pd(0)中心,防止各Pd(0)靠近形成无活性的钯黑,提升了催化剂的稳定性[34];新生的富电子cis-(PR3)2-Pd(0)的亲核性强,更容易进攻并插入C—X键,提高氧化加成步骤的速率,在中间体R-Pd(Ⅱ)-R′(R:a1~a4,R′:b1~b4)中,2个较大R、R′基团和2个大位阻的PR3配体拥挤在Pd(0)中心周围,产生巨大的空间张力,为了缓解这种张力,R与R′成键并离开变得容易,加速了偶联产物R-R′的生成和cis-(PR3)2-Pd(0)的再生[35-36]。
催化反应后,cis-(PR3)2-Pd(0)在回收过程中被O2等氧化剂氧化为Pd(Ⅱ),与体系中离子交换所得大量溴离子(Br-)结合,首先得到cis-[Pd(Xantphos)Br2] 和cis-[Pd(PPh3)2Br2],Xantphos的刚性骨架维持[Pd(Xantphos)Br2]的顺式结构,cis-[Pd(PPh3)2Br2]则翻转为热力学稳定的trans-[Pd(PR3)2Br2]。
本研究通过实验与理论计算相结合的方法,探究了PR3调控下[Pd(COD)X2](X=Cl、Br)的Pd(Ⅱ)中心结构与催化性能的关系。结果表明,配体PR3能显著提升C—C偶联反应催化效率:[Pd(COD)X2]/Xantphos > [Pd(COD)X2]/PPh3 > 市售trans-[Pd(PPh3)2Cl2] > [Pd(COD)X2],溴代前驱体([Pd(COD)Br2])的催化性能优于氯代前驱体([Pd(COD)Cl2])。其核心在于PR3的构型调控与卤素键强协同影响Pd(0)生成:PR3通过富电子的膦与Pd(0)配位,PR3的大位阻空间效应抑制Pd(0)团聚失活;不添加PR3配体时,新生的Pd(0)因高表面活性快速团聚,使收率极低且不可逆;市售催化剂具有稳定配位结构,性能稳定但无显著优势。机理研究表明,Xantphos的刚性骨架延续钯中心的配位,得到的cis-(Xantphos)-Pd(0)暴露更多活性位点;PPh3取代的cis-Pd(PPh3)2X2易翻转为反式构型,为cis-(PPh3)2-Pd(0)与trans-(PPh3)2-Pd(0)混合物;此外,Pd—Br键能较低,更易断裂,可更快生成Pd(0),故[Pd(COD)Br2]收率高于[Pd(COD)Cl2]。
ZHANG L X, ZHOU F B, CHENG G Y, YANG X, XIAO D D, JIANG D H, CHENG H C, YUAN B X, SHI J C. Synthesis and characterization of terphenyl phosphine ligands and their palladium catalyst precursors[J]. Organometallics, 2025, 44(15): 1653-1659 doi: 10.1021/acs.organomet.5c00129
WANG Z, ZHANG Z G, LU X Y. Effect of halide ligands on the reactivity of carbon-palladium bonds: Implications for designing catalytic reactions[J]. Organometallics, 2000, 19(5): 775-780 doi: 10.1021/om990670h
XIE H J, FAN T, LEI Q F, FANG W J. New progress in theoretical studies on palladium-catalyzed C—C bond-forming reaction mechanisms[J]. Sci. China Chem., 2016, 59(11): 1432-1447 doi: 10.1007/s11426-016-0018-2
ZHAN B B, JIN L, SHI B F. Palladium-catalyzed enantioselective C—H functionalization via C—H palladation[J]. Trends Chem., 2022, 4(3): 220-235 doi: 10.1016/j.trechm.2021.12.005
RAGO A J, DONG G. Synthesis of indoles, indolines, and carbazoles via palladium-catalyzed C—H activation[J]. Green Synth. Catal., 2021, 2(2): 216-227
TRZECIAK A M, WOJTKÓW W, ZIÓŁKOWSKI J J. Catalytic activity of palladium complexes, PdCl2(COD) and PdCl2(P(OPh)3)2, in methoxy-carbonylation of iodobenzene[J]. Inorg. Chem. Commun., 2003, 6(7): 823-826 doi: 10.1016/S1387-7003(03)00105-9
MOHANTY S, PUNJI B, BALAKRISHNA M S. Synthesis and reaction kinetics of Pd(1, 5-cyclooctadiene)Cl2 with N, N′-methylene-bis (2-aminopyridyl): An efficient catalyst for Suzuki-cross-coupling reactions[J]. Polyhedron, 2006, 25(3): 815-820 doi: 10.1016/j.poly.2005.08.013
FU G C. The development of versatile methods for palladium-catalyzed coupling reactions of aryl electrophiles through the use of P(t-Bu)3 and PCy3 as ligands[J]. Accounts Chem. Res., 2008, 41(11): 1555-1564 doi: 10.1021/ar800148f
FLECKENSTEIN C A, PLENIO H. Sterically demanding trialkylphosphines for palladium-catalyzed cross coupling reactions-alternatives to P(t-Bu)3[J]. Chem. Soc. Rev., 2010, 39(2): 694-711 doi: 10.1039/B903646F
WANG J R, XU B, WANG Y B, XIA G Z, ZHANG Z M, ZHANG J L. Pd-catalyzed enantioselective three-component carboamination of 1, 3-cyclohexadiene[J]. J. Am. Chem. Soc., 2024, 146(31): 21231-21238 doi: 10.1021/jacs.4c07382
SEECHURN C C C J, SPERGER T, SCRASE T G, SCHOENEBECK F, COLACOT T J. Understanding the unusual reduction mechanism of Pd(Ⅱ) to Pd(Ⅰ): Uncovering hidden species and implications in catalytic cross-coupling reactions[J]. J. Am. Chem. Soc., 2017, 139(14): 5194-5200 doi: 10.1021/jacs.7b01110
HUPPERICH D, PONCE-DE-LEÓN J, FUNES-ARDOIZ I, SPERGER T, SCHOENEBECKET F. Sterically induced acceleration of aryl halide activation by Pd(0): A radical alternative to 2-electron oxidative addition[J]. J. Am. Chem. Soc., 2025, 147(23): 19941-19948 doi: 10.1021/jacs.5c04407
SOLIN N, SZABÓ K J. Mechanism of the η3-η1-η3 isomerization in allylpalladium complexes: Solvent coordination, ligand, and substituent effects[J]. Organometallics, 2001, 20(25): 5464-5471 doi: 10.1021/om010793d
DEANGELIS A J, GILDNER P G, CHOW R, COLACOT T J. Generating active "L-Pd (0)" via neutral or cationic π-allylpalladium complexes featuring biaryl/bipyrazolylphosphines: Synthetic, mechanistic, and structure-activity studies in challenging cross-coupling reactions[J]. J. Org. Chem., 2015, 80(13): 6794-6813. doi: 10.1021/acs.joc.5b01005
Shaughnessy K H. Development of palladium precatalysts that efficiently generate LPd(0) active species[J]. Isr. J. Chem., 2020, 60(3/4): 180-194
RETTIG M F, WING R M, WIGER G R. X-ray crystallographic, chemical, and spectroscopic studies of the palladium dichloride complexes of cyclonona-1, 5-diene, cycloocta-1, 5-diene, cycloocta-1, 4-diene, and cyclohepta-1, 4-diene[J]. J. Am. Chem. Soc., 1981, 103(11): 2980-2986 doi: 10.1021/ja00401a012
KLEIN A, DOGAN A, FETH M, BERTAGNOLLI H. Cyclooctadi-enemethylpalladium complexes: Synthesis, structures and reactivity[J]. Inorg. Chim. Acta, 2003, 343: 189-201 doi: 10.1016/S0020-1693(02)01231-8
STOCKLAND JR R A, ANDERSON G K, RATH N P, BRADDOCK W J, ELLEGOOD J C. Synthesis of the complexes [PdClR (cod)](R=benzyl, ethyl; cod=1, 5-cyclooctadiene). β-Elimination from [PdClEt(cod)] to give the η1, η2 and η3 isomers of [Pd2(μ-Cl)2(C8H13)2][J]. Can. J. Chem., 1996, 74(11): 1990-1997 doi: 10.1139/v96-226
POWELL D B, LEEDHAM T J. Infrared and Raman spectra of 1, 5-cyclooctadiene complexes of rhodium, palladium and platinum[J]. Spectroc. Acta Pt. A‒Molec. Biomolec. Spectr., 1972, 28(2): 337-341
KUNKELY H, VOGLER A. Electronic spectrum and photoreactivity of dichloro (1, 5-cyclooctadiene)-palladium(Ⅱ)[J]. J. Organomet. Chem., 1998, 559(1/2): 223-225
NEESE F. Software update: The ORCA program system, version 4.0[J]. Wiley Interdiscip. Rev. ‒Comput. Mol. Sci., 2018, 8(1): e1327 doi: 10.1002/wcms.1327
BECKE A D. Density-functional thermochemistry. Ⅲ. The role of exact exchange[J]. J. Chem. Phys., 1993, 98(7): 5648-5652 doi: 10.1063/1.464913
ARAVENA D, NEESE F, PANTAZIS D A. Improved segmented all-electron relativistically contracted basis sets for the lanthanides[J]. J. Chem. Theory Comput., 2016, 12(3): 1148-1156 doi: 10.1021/acs.jctc.5b01048
ŠTĚPNIČKA P, CÍSAŘOVÁ I. Crystal structure of dibromo (η4-1, 5-cyclooctadiene) palladium(Ⅱ)[J]. Collect. Czech. Chem. Commun., 1996, 61(9): 1335-1341 doi: 10.1135/cccc19961335
MOHANTY S, PUNJI B, BALAKRISHNA M S. Synthesis and reaction kinetics of Pd(1, 5-cyclooctadiene)Cl2 with N, N′-methylene-bis (2-aminopyridyl): An efficient catalyst for Suzuki-cross-coupling reactions[J]. Polyhedron, 2006, 25(3): 815-820 doi: 10.1016/j.poly.2005.08.013
STEPHENSON M D, HARDIE M J. Extended structures of transition metal complexes of 6, 7-dicyanodipyridoquinoxaline: π-Stacking, weak hydrogen bonding, and CN-π interactions[J]. Cryst. Growth Des., 2006, 6(2): 423-432 doi: 10.1021/cg050366t
MAGISTRATO A, PREGOSIN P S, ALBINATI A, ROTHLISBERGER U. The role of π-π stacking interactions in square planar palladium complexes. combined quantum mechanics/molecular mechanics QM/MM studies[J]. Organometallics, 2001, 20(20): 4178-4184 doi: 10.1021/om010485f
DATSENKO V P, NELYUBINA Y V, SMOL′YAKOV A F, LOGINOV D A. Cyclooctadiene iridium complexes [Cp*Ir(COD)X]+ (X=Cl, Br, I): Synthesis and application for oxidative coupling of benzoic acid with alkynes[J]. J. Organomet. Chem., 2018, 874: 7-12 doi: 10.1016/j.jorganchem.2018.08.014
HARTLEY F R. The cis- and trans-effects of ligands[J]. Chem. Soc. Rev., 1973, 2(2): 163-179 doi: 10.1039/cs9730200163
D′ALTERIO M C, CASALS-CRUAÑAS È, TZOURAS N V, TALARICO G, NOLAN S P, POATER A. Mechanistic aspects of the palladium-catalyzed Suzuki-Miyaura cross-coupling reaction[J]. Chem. ‒Eur. J., 2021, 27(54): 13481-13493
JEDDI N, SCOTT N W J, FAIRLAMB I J S. Well-defined Pdn clusters for cross-coupling and hydrogenation catalysis: New opportunities for catalyst design[J]. ACS Catal., 2022, 12(19): 11615-11638 doi: 10.1021/acscatal.2c03345
SIVENDRAN N, PIRKL N, HU Z Y, DOPPIU A, GOOSSEN L J. Halogen-bridged methylnaphthyl palladium dimers as versatile catalyst precursors in coupling reactions[J]. Angew. Chem. ‒Int. Edit., 2021, 60(47): 25151-25160 doi: 10.1002/anie.202110450
CASADO A L, ESPINET P. On the configuration resulting from oxidative addition of RX to Pd(PPh3)4 and the mechanism of the cis-to-trans isomerization of [PdRX(PPh3)2] complexes (R=aryl, X=halide)[J]. Organometallics, 1998, 17(5): 954-959 doi: 10.1021/om9709502
PORTNOY M, MILSTEIN D. Chelate effect on the structure and reactivity of electron-rich palladium complexes and its relevance to catalysis[J]. Organometallics, 1993, 12(5): 1655-1664 doi: 10.1021/om00029a025
TOLMAN C A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis[J]. Chem. Rev., 1977, 77(3): 313-348
TATSUMI K, HOFFMANN R, YAMAMOTO A, STILLE J K. Reductive elimination of d8-organotransition metal complexes[J]. Bull. Chem. Soc. Jpn., 1981, 54(6): 1857-1867 doi: 10.1246/bcsj.54.1857

图 3 前驱体在THF中不同时间下的紫外可见光谱图
Figure 3 UV-Vis spectra of the precursors in THF at different times

图 4 前驱体在DCM中不同时间下的紫外可见光谱图
Figure 4 UV-Vis spectra of the precursors in DCM at different times

图 6 trans-[Pd(PPh3)2Br2] (a)和cis-[Pd(Xantphos)Br2] (b)的椭球率30%的晶体结构
Figure 6 Crystal structure of trans-[Pd(PPh3)2Br2] (a) and cis-[Pd(Xantphos)Br2] (b) with 30% probability ellipsoids
Symmetry code: ⅰ 1-x, 1-y, 1-z.

图 7 trans-[Pd(PPh3)2Br2] (a)和cis-[Pd(Xantphos)Br2] (b)的分子堆积图中的氢键
Figure 7 Hydrogen bonds in the molecular stacking diagrams of trans-[Pd(PPh3)2Br2] (a) and cis-[Pd(Xantphos)Br2] (b)
Symmetry codes: ⅰ 1-x, 1-y, 1-z; ⅱ 1/2-x, 1/2+y, z; ⅲ-1/2+x, 3/2-y, 1-z; ⅳ-1/2+x, 1/2-y, 1-z; ⅴ 1/2-x, -1/2+y, z for trans-[Pd(PPh3)2Br2]; ⅰ 1-x, 2-y, 1-z; ⅱ x, y, -1+z for cis-[Pd(Xantphos)Br2].

图 8 C—H…π作用的平面和质心: (a) trans-[Pd(PPh3)2Br2]、(b) cis-[Pd(Xantphos)Br2]
Figure 8 Planes and centroids for the C—H…π interactions: (a) trans-[Pd(PPh3)2Br2], (b) cis-[Pd(Xantphos)Br2]
Symmetry codes: ⅱ 1/2-x, 1/2+y, z; ⅳ-1/2+x, 1/2-y, 1-z for trans-[Pd(PPh3)2Br2]; ⅰ 1-x, 2-y, 1-z; ⅱ x, y, -1+z for cis-[Pd(Xantphos)Br2].

图 9 [Pd(COD)X2]催化Suzuki反应的机理
Figure 9 Mechanism of the Suzuki reaction catalyzed by [Pd(COD)X2]
表 1 trans-[Pd(PPh3)2Br2]和cis-[Pd(Xantphos)Br2]的晶体学数据
Table 1. Crystallographic data for trans-[Pd(PPh3)2Br2] and cis-[Pd(Xantphos)Br2]
| Parameter | trans-[Pd(PPh3)2Br2] | cis-[Pd(Xantphos)Br2] |
| Empirical formula | C36H30Br2P2Pd·2CH2Cl2 | C39H32Br2OP2Pd·C4H8O2 |
| Formula weight | 960.61 | 932.91 |
| Crystal system | Orthorhombic | Triclinic |
| Space group | Pbca | P1 |
| a / nm | 2.017 58(9) | 1.032 33(4) |
| b / nm | 0.807 92(3) | 1.143 37(5) |
| c / nm | 2.322 16(9) | 1.722 37(7) |
| α / (°) | 79.611(2) | |
| β / (°) | 87.514(2) | |
| γ / (°) | 83.738(2) | |
| Volume / nm3 | 3.785 2(3) | 1.987 13(14) |
| Z | 4 | 2 |
| Dc / (Mg·m-3) | 1.686 | 1.559 |
| μ / mm-1 | 10.035 | 7.183 |
| F(000) | 1 904 | 936 |
| θ range / (°) | 4.38-68.95 | 2.61-68.66 |
| Index ranges | -24 ≤ h ≤ 22, -9 ≤ k ≤ 9, -27 ≤ l ≤ 27 | -12 ≤ h ≤ 12, -13 ≤ k ≤ 13, -17 ≤ l ≤ 20 |
| Reflection collected | 23 098 | 28 080 |
| Independent reflection | 3 480 (Rint=0.107 0) | 7 268 (Rint=0.076 8) |
| Goodness-of-fit on F 2 | 1.078 | 1.070 |
| Final R indices [I > 2σ(I)] | R1=0.035 4, wR2=0.085 5 | R1=0.052 4, wR2=0.151 8 |
| R indices (all data) | R1=0.052 7, wR2=0.090 7 | R1=0.066 2, wR2=0.166 0 |
| Largest diff. peak and hole / (e·nm-3) | 531 and -569 | 1 088 and -1 052 |
 下载: 导出CSV
下载: 导出CSV
表 2 前驱体中Pd—X键长的实验值和理论值
Table 2. Experimental and theoretical values of Pd—X bonds of the precursors
| Precursor | Bond | Bond length / nm | |
| Experimental | Theoretical | ||
| [Pd(COD)Cl2] | Pd—Cl1 | 0.230 3 | 0.231 5 |
| Pd—Cl2 | 0.230 9 | 0.229 2 | |
| [Pd(COD)Br2] | Pd—Br1 | 0.242 5 | 0.241 8 |
| Pd—Br2 | 0.244 3 | 0.243 2 | |
下载: 导出CSV
表 3 前驱体的前线轨道能级
Table 3. Frontier orbital energy levels of the precursors
| Precursor | E / eV | Bandgap (ΔE) / eV | |
| HOMO | LUMO | ||
| [Pd(COD)Cl2] | -6.844 | -2.852 | 3.992 |
| [Pd(COD)Br2] | -6.343 | -2.950 | 3.392 |
下载: 导出CSV
表 4 [Pd(COD)X2]在不同底物的Suzuki偶联反应中的催化活性(反应Ⅰ)
Table 4. Catalytic activity of [Pd(COD)X2] in Suzuki coupling reactions with different substrates (Reaction Ⅰ)*
|
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 79 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 10 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 1.5 | 82 |
| 4 | [Pd(COD)Cl2] | Xantphos | 1.5 | 94 |
| 5 | [Pd(COD)Br2] | 12.0 | 15 | |
| 6 | [Pd(COD)Br2] | PPh3 | 1.5 | 88 |
| 7 | [Pd(COD)Br2] | Xantphos | 1.5 | 97 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载: 导出CSV
表 5 [Pd(COD)X2]在不同底物的Suzuki偶联反应中的催化活性(反应Ⅱ)
Table 5. Catalytic activity of [Pd(COD)X2] in Suzuki coupling reactions with different substrates (Reaction Ⅱ)*
|
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 61 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 10 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 5.0 | 83 |
| 4 | [Pd(COD)Cl2] | Xantphos | 3.0 | 95 |
| 5 | [Pd(COD)Br2] | 12.0 | 15 | |
| 6 | [Pd(COD)Br2] | PPh3 | 5.0 | 84 |
| 7 | [Pd(COD)Br2] | Xantphos | 3.0 | 97 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载: 导出CSV
表 6 [Pd(COD)X2]在不同底物的Suzuki偶联反应中的催化活性(反应Ⅲ)
Table 6. Catalytic activity of [Pd(COD)X2] in Suzuki coupling reactions with different substrates (Reaction Ⅲ)*
|
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 73 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 16 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 5.0 | 80 |
| 4 | [Pd(COD)Cl2] | Xantphos | 3.0 | 92 |
| 5 | [Pd(COD)Br2] | 12.0 | 20 | |
| 6 | [Pd(COD)Br2] | PPh3 | 5.0 | 83 |
| 7 | [Pd(COD)Br2] | Xantphos | 3.0 | 92 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载: 导出CSV
表 7 [Pd(COD)X2]在不同底物的Suzuki偶联反应中的催化活性(反应Ⅳ)
Table 7. Catalytic activity of [Pd(COD)X2] in Suzuki coupling reactions with different substrates (Reaction Ⅳ)*
|
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 77 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 15 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 5.0 | 79 |
| 4 | [Pd(COD)Cl2] | Xantphos | 3.0 | 94 |
| 5 | [Pd(COD)Br2] | 12.0 | 23 | |
| 6 | [Pd(COD)Br2] | PPh3 | 5.0 | 81 |
| 7 | [Pd(COD)Br2] | Xantphos | 3.0 | 96 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载: 导出CSV
表 8 [Pd(COD)X2]在不同底物的Suzuki偶联反应中的催化活性(反应Ⅴ)
Table 8. Catalytic activity of [Pd(COD)X2] in Suzuki coupling reactions with different substrates (Reaction Ⅴ)*
|
||||
| Entry | Catalyst | PR3 | Time / h | Yield / % |
| 1 | trans-[Pd(PPh3)2Cl2] | 4.0 | 80 | |
| 2 | [Pd(COD)Cl2] | 12.0 | 21 | |
| 3 | [Pd(COD)Cl2] | PPh3 | 5.0 | 83 |
| 4 | [Pd(COD)Br2] | 12.0 | 32 | |
| 5 | [Pd(COD)Br2] | PPh3 | 5.0 | 89 |
| * Unless otherwise specified, the reaction system comprised bromopyridine derivatives (5.0 mmol), phenylboronic acid derivatives (1.1 equivalents), Na2CO3 (2.0 equivalents), Pd catalyst (x=1.0%), and PR3 (x=1%-2%) in distilled THF (50.0 mL)/H2O (13.0 mL) under argon atmosphere at 90 ℃ for 1.5-12.0 h, and the product was isolated by column chromatography. | ||||
下载: 导出CSV
表 9 Suzuki反应后回收的催化剂的主要键长(nm)和键角(°)
Table 9. Main bond lengths (nm) and bond angles (°) of the catalysts recovered after the Suzuki reaction
| trans-[Pd(PPh3)2Br2] | |||||
| Pd—Br1 | 0.242 62 | Pd—P1 | 0.232 65 | P1—C1 | 0.181 34 |
| P1—C13 | 0.182 52 | ||||
| Br1—Pd1—P1 | 92.631 | Pd1—P1—C1 | 161.839 | C1—P1—C7 | 93.968 |
| C7—P1—C13 | 161.328 | ||||
| cis-[Pd(Xantphos)Br2] | |||||
| Pd—Br1 | 0.220 38 | Pd—Br2 | 0.222 57 | Pd—P1 | 0.222 39 |
| Pd—P2 | 0.217 69 | P1—C1 | 0.242 58 | P1—C8 | 0.244 30 |
| P1—Pd—Br2 | 85.867 | P2—Pd—Br1 | 84.934 | P1—Pd—P2 | 100.757 |
| Br2—Pd—Br1 | 86.521 | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: