Received Date:
15 June 2025 Revised Date:
22 May 2026 Available Online:
10 July 2026
Abstract:
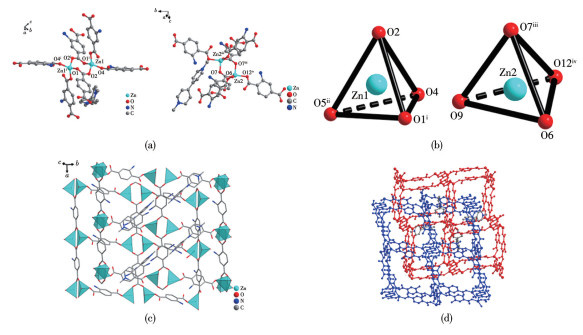
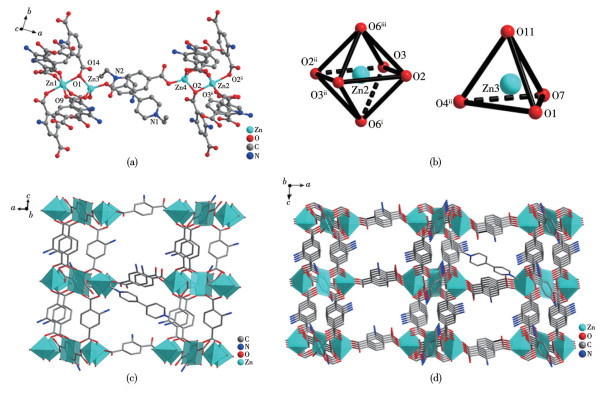
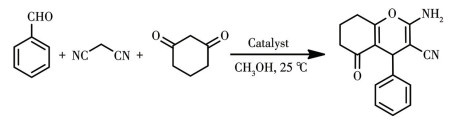
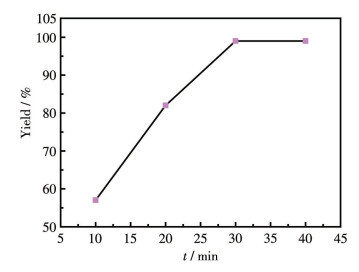
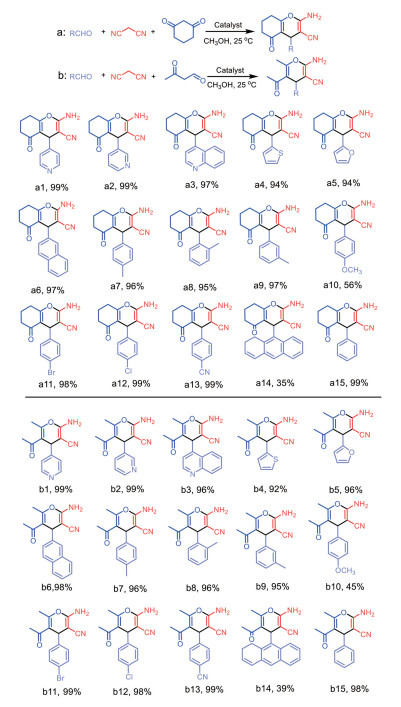
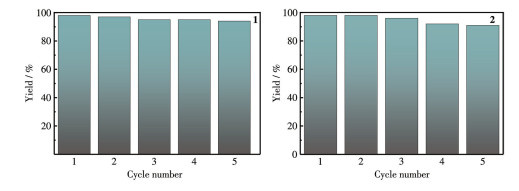
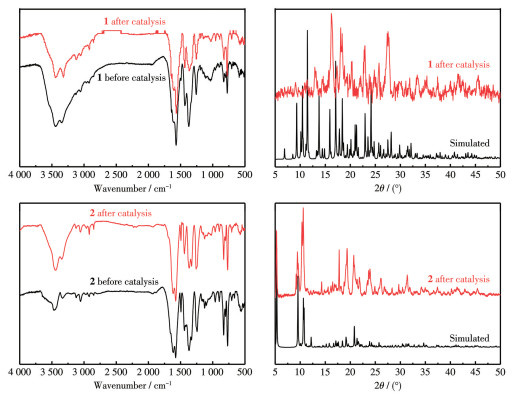
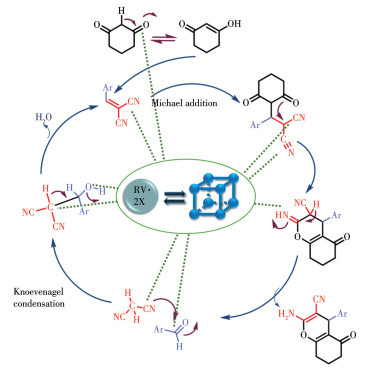
By virtue of the solvothermal synthetic method, 1,1′-dimethyl-4,4′-bipyridinium cation (methyl viologen cation, MV2+) and 1,1′-diethyl-4,4′-bipyridinium cation (ethyl viologen cation, EV2+) were embedded into the framework of Zn-NH2-BDC, achieving two viologen-modified metal-organic frameworks: {(MV)[Zn2(NH2-BDC)3]}n (1), {(EV[Zn3(NH2-BDC)4]}n (2) (NH2-H2BDC=2-aminoterephthalic acid). The two complexes have shown considerable catalytic activities under mild conditions, namely, they could be used as efficient and recyclable catalysts for the Knoevenagel condensation-Michael addition cyclization reaction, and consequently, to obtain 4H-pyran derivatives, and the yield could reach over 98%. Remarkably, two catalysts could be reused at least five times while maintaining their catalytic activities.
Supporting information is available at http://www.wjhxxb.cn
[1]
SKOMMER J, WLODKOWIC D, MÄTTÖ M, ERAY M, PELKONEN J. HA14-1, a small molecule Bcl-2 antagonist, induces apoptosis and modulates action of selected anticancer drugs in follicular lymphoma B cells[J]. Leuk. Res., 2006, 30(3): 322-331 doi: 10.1016/j.leukres.2005.08.022
[2]
NARAJJI C, KARVEKAR M D, DAS A K. Synthesis and antioxidant activity of 1,2-bis(1-ethyl-2-substitutedphenylindolizin-3-yl)diselane[J]. Asian J. Chem., 2008, 20(8): 6183-6188
[3]
MOHR S J, CHIRIGOS M A, FUHRMAN F S, PRYOR J W. Pyran copolymer as an effective adjuvant to chemotherapy against a murine leukemia and solid tumor[J]. Cancer Res., 1975, 35(12): 3750-3754
[4]
SON H D, THI THU HA N, TIEN TUNG D, THI KIM GIANG N. Synthesis, biological evaluation and induced fit docking simulation study of D-glucose-conjugated 1H-1,2,3-triazoles having 4H-pyrano[2,3-d]pyrimidine ring as potential agents against bacteria and fungi[J]. New J. Chem, 2022, 46(17): 8303-8323 doi: 10.1039/D1NJ05330B
[5]
WANG H J, LU J, ZHANG Z H. Highly efficient three-component, one-pot synthesis of dihydropyrano[3,2-c]chromene derivatives[J]. Monatsh. Chem., 2010, 141(10): 1107-1112 doi: 10.1007/s00706-010-0383-4
[6]
LIU X Y, WU W, LI X J, WANG C Y, CHAI K, YUAN F R, ZHENG H J. The compound (E)-2-(3,4-dihydroxystyryl)-3-hydroxy-4H-pyran-4-one alleviates neuroinflammation and cognitive impairment in a mouse model of Alzheimer′s disease[J]. Neural Regen. Res., 2025, 20(11): 3330-44 doi: 10.4103/NRR.NRR-D-23-01890
[7]
DU F Y, ZHOU Q F, FU X X, ZHENG Y J, FANG W H, YANG J Y, CHEN G L. Synthesis and biological evaluation of 2,2-dimethylbenzopyran derivatives as potent neuroprotection agents[J]. RSC Adv., 2019, 9(5): 2498-2508 doi: 10.1039/C8RA10424G
[8]
SHUKLA S K, MISHRA A K, AROTIBA O A, MAMBA B B. Chitosan-based nanomaterials: A state‑of‑the‑art review[J]. Int. J. Biol. Macromol, 2013, 59: 46-58 doi: 10.1016/j.ijbiomac.2013.04.043
[9]
GUTIERREZ L F, NOPE E, ROJAS H A, CUBILLOS J A, SATHICQ A G, ROMANELLI G P, MARTÍNEZ J J. New application of decaniobate salt as basic solid in the synthesis of 4H-pyrans by microwave assisted multicomponent reactions[J]. Res. Chem. Int., 2018, 44(9): 5559-5568 doi: 10.1007/s11164-018-3440-y
[10]
ELNAGDI N M H, AL-HOKBANY N S. Organocatalysis in synthesis: L-proline as an enantioselective catalyst in the synthesis of pyrans and thiopyrans[J]. Molecules, 2012, 17(4): 4300-4312 doi: 10.3390/molecules17044300
[11]
VALKENBERG M H, HÖLDERICH W F. Preparation and use of hybrid organic-inorganic catalysts[J]. Catal. Rev., 2002, 44(2): 321-374 doi: 10.1081/CR-120003497
[12]
SOLIMAN A A, MOHAMED G G, HOSNY W M, ELMAWGOOD M A. Synthesis, spectroscopic and thermal characterization of new sulfasalazine metal complexes[J]. Synth. React. Inorg. Met. ‒Org. Nano-Metal Chem., 2005, 35(6): 483-490 doi: 10.1081/SIM-200067043
[13]
AKHLAGHI Z, NAIMI-JAMAL M R, PANAHI L, DEKAMIN M G. Solvent-free mechanochemical multicomponent preparation of 4H-pyrans catalyzed by Cu2(NH2-BDC)2(DABCO) metal-organic framework[J]. Heliyon, 2023, 9(2): e13522 doi: 10.1016/j.heliyon.2023.e13522
[14]
YANG K W, JIANG J W. Transforming CO2 into methanol with N-heterocyclic carbene-stabilized coinage metal hydrides immobilized in a metal-organic framework UiO-68[J]. ACS Appl. Mater. Interfaces, 2021, 13(49): 58723-58736 doi: 10.1021/acsami.1c18885
[15]
ZHANG Y, YANG X, ZHOU H C. Synthesis of MOFs for heterogeneous catalysis via linker design[J]. Polyhedron, 2018, 154: 189-201 doi: 10.1016/j.poly.2018.07.021
[16]
WANG P, LI X H, ZHANG P, ZHANG X F, SHEN Y, ZHENG B, WU J S, LI S, FU Y, ZHANG W N, HUO F W. Transitional MOFs: Exposing metal sites with porosity for enhancing catalytic reaction performance[J]. ACS Appl. Mater. Interfaces, 2020, 12(21): 23968-23975 doi: 10.1021/acsami.0c04606
[17]
LIU J W, CHEN L F, CUI H, ZHANG J Y, ZHANG L, SU C Y. Applications of metal-organic frameworks in heterogeneous supramolecular catalysis[J]. Chem. Soc. Rev., 2014,43(16): 6011-6061 doi: 10.1039/C4CS00094C
[18]
LI T, LI N, SHI D. A highly efficient and recyclable CuI@UiO-67-bpy catalyst for direct sp2 C—H arylation of azoles[J]. New J. Chem., 2024,48(46): 19418-19426 doi: 10.1039/D4NJ03726J
[19]
SAGARA T, TAHARA H. Redox of viologen for powering and coloring[J]. Chem. Rec., 2021,21(9): 2375-2388 doi: 10.1002/tcr.202100082
[20]
WANG M S, XU G, ZHANG Z J, GUO G C. Inorganic-organic hybrid photochromic materials[J]. Chem. Commun., 2010, 46(3): 361-376 doi: 10.1039/B917890B
[21]
LI P X, WANG M S, CAI L Z, WANG G E, GUO G C. Rare electron-transfer photochromic and thermochromic difunctional compounds[J]. J. Mater. Chem. C, 2015, 3(2): 253-256 doi: 10.1039/C4TC01315H
[22]
SUN Y H, LI C L, WANG W F, WANG S H, LI P X, GUO G C. A photochromic and scintillation Eu-MOF with visual X-ray detection in bright and dark environments[J]. Chem. Commun., 2022, 58(25): 4056-4059 doi: 10.1039/D2CC00166G
[23]
PAN Q Y, SUN M E, ZHANG C, LI L K, LIU H L, LI K J, LI H Y, ZANG S Q. A multi-responsive indium-viologen hybrid with ultrafast-response photochromism and electrochromism[J]. Chem. Commun., 2021, 57(86): 11394-11397 doi: 10.1039/D1CC05070B
[24]
LI H Y, HUA X, FU T, LIU X F, ZANG S Q. Photochromic and electrochromic properties of a viologen-based multifunctional Cd-MOF[J]. Chem. Commun., 2022, 58(56): 7753-7756 doi: 10.1039/D2CC02703H
[25]
LUO Y, YING S W, LI S J, LI L K, LI H Y, ASAD M, ZANG S Q, MAK T C W. Photo/electrochromic dual responsive behavior of a cage-like Zr-viologen metal-organic polyhedron (MOP)[J]. Inorg. Chem., 2022, 61(6): 2813-2823 doi: 10.1021/acs.inorgchem.1c03203
[26]
WANG T, ZHANG L, LIU J, LI X X, YUAN L, LI S L, LAN Y Q. A viologen-functionalized metal-organic framework for efficient CO2 photoreduction reaction[J]. Chem. Commun., 2022, 58(54): 7507-7510 doi: 10.1039/D2CC02650C
[27]
SHELDRICK G. Shelxt-integrated space-group and crystal-structure determination[J]. Acta Crystallogr. Sect. A, 2015, A71(1): 3-8
[28]
THALLAPALLY P K, NANGIA A. A Cambridge structural database analysis of the $\mathrm{C}—\mathrm{H} \cdots \mathrm{Cl} $ interaction: $\mathrm{C}—\mathrm{H} \cdots \mathrm{Cl}— $ and $\mathrm{C}—\mathrm{H} \cdots \mathrm{Cl}—\mathrm{M} $ often behave as hydrogen bonds but $\mathrm{C}—\mathrm{H} \cdots \mathrm{Cl}—\mathrm{C} $ is generally a van der Waals interaction[J]. CrystEngComm, 2001,3(27): 114-119 doi: 10.1039/B102780H
[29]
CHENG L M, ZHANG J Y, ZHAN C, XU H, GONG C H, XIE J L. Metal-organic frameworks constructed using acid-base mixed ligands, carboxylic acids and N-containing chalcone, and their catalytic performance for Knoevenagel condensation[J]. New J. Chem., 2024,48(19): 8597-8602 doi: 10.1039/D3NJ05164A
[30]
PARK K K, LEE C W, OH S Y, PARK J W. Viologen-mediated reductive dehalogenation of α-halogeno ketones[J]. J. Chem. Soc. Perkin Trans. 1, 1990(8): 2356-2357
[31]
JUSTIN A, ESPÍN J, KOCHETYGOV I, ASGARI M, TRUKHINA O, QUEEN W L. A two step postsynthetic modification strategy: Appending short chain polyamines to Zn‑NH2‑BDC MOF for Enhanced CO2 adsorption[J]. Inorg. Chem., 2021, 60(16): 11720-11729 doi: 10.1021/acs.inorgchem.1c01216
[32]
CHENG X, GUO L, WANG H, GU J, YANG Y, KIRILLOVA M V, KIRILLOV A M. Coordination polymers constructed from an adaptable pyridine-dicarboxylic acid linker: Assembly, diversity of structures, and catalysis[J]. Inorg. Chem., 2022, 61(45): 17951-17962 doi: 10.1021/acs.inorgchem.2c01855
[33]
ROSTAMIZADEH S, DANESHFAR Z, KHAZAEI A. Ferric sulfasalazine sulfa drug complex supported on cobalt ferrite cellulose; evaluation of its activity in MCRs[J]. Catal. Lett., 2020, 150(7): 2091-2114 doi: 10.1007/s10562-020-03101-6

 下载:
下载:

 下载:
下载:










