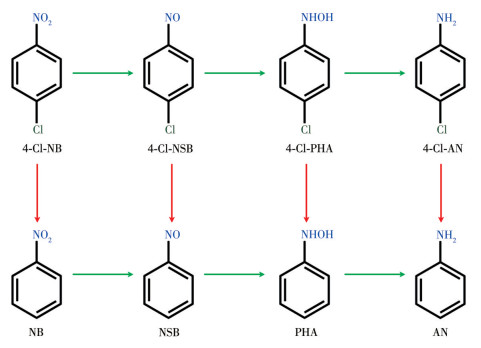
Figure 1.
Reaction network of the hydrogenation of halogenated nitroaromatics, exemplified by 4-chloronitrobenzene (4-Cl-NB)

Accelerating catalysts with enhanced activity and selectivity can minimize or eliminate the formation of by‑products, thereby aligning with the principles of energy conservation, emission reduction, and sustainable development[1]. However, when reactions involve competitive pathways that result in the generation of multiple intermediates or products, achieving high selectivity for the target product becomes crucial and difficult[2-4]. One typical example is the selective hydrogenation of nitroaromatics to functional amines[5], such as the selective hydrogenation of halogenated nitroaromatics to halogenated aromatic amines which are important chemical raw materials and intermediates for the synthesis of pharmaceuticals, dyes, and pesticides[6]. However, the complex hydrogenation process occurs through both direct and condensation routes, with the formation of up to five intermediates (nitroso, hydroxylamine, azoxy, azo, and hydrazo) together with the desired amine product[7]. Furthermore, it entails a competitive reaction of dehalogenation in the hydrogenation, which results in the formation of dehalogenated by-products (Fig. 1). This not only reduces the purity of the product but also generates strong acids (HX, where X=F, Cl, Br, I). These acids can cause severe corrosion of the reactor, posing a significant safety hazard. The highly selective hydrogenation of halogenated nitroaromatics is thus both essential and challenging for obtaining the desired amine products.
In general, the selective catalytic hydrogenation of halogenated nitroaromatics can be enhanced by modulating the structure of the catalyst active site, as this allows to regulation of the adsorption energy and conformation of the reactants/intermediates[8-10]. One of the most prevalent strategies is the introduction of organic dehalogenation inhibitors by modifying the metal surface with organic ligands. The ligands typically comprise coordination atoms, such as P, S, N, and so forth[11-15]. The steric or electronic effects generated by the organic ligands can effectively manipulate the structure and strength of the interaction between the substrate and the active site, thereby achieving high selectivity[16-19]. However, organic ligands on metal surfaces have been observed to significantly impair or even deactivate the catalyst in numerous instances[20-21].
Furthermore, halogens act as electron-withdrawing groups, which frequently result in a notable accumulation of hydroxylamines. The hydrogenation of hydroxylamine to aniline occurs at a relatively slow rate, which further leads to a reduction in the overall reaction rate[22]. The hydroxylamine accumulation also serves to enhance the extent of the condensation. The formation of azo compounds not only slows the reaction rate but also negatively affects the product quality, as azo compounds possess a particularly dark color[2]. The introduction of vanadium species has been documented as an effective strategy to resolve the hydroxylamine accumulation issue, as a catalytic amount of vanadium enables the rapid conversion of hydroxylamine to amine via disproportionation[2, 22-23]. Therefore, it is expected that the selective catalytic hydrogenation of halogenated nitroaromatics to their corresponding halogenated aromatic amines can be achieved by effectively combining the benefits of both organic and inorganic modifications while minimizing their respective drawbacks.
Herein, we achieved highly selective catalytic hydrogenation of halogenated nitroaromatics by employing a strategy of co-modification of the Pd/C catalyst with both organic ligand (PPh3) and inorganic species (NaVO3). The catalyst applied was a heterogeneous Pd/C catalyst with Pd nanoparticles (NPs) supported on activated carbon. The Pd NPs contain high-valence Pd species at the metal-support interface and zero-valence Pd species on the metal surface. A systematic investigation demonstrated that the coordination of PPh3 on the zero-valence Pd sites inhibited the coplanar adsorption of halogenated nitroaromatics on Pd, thereby inhibiting dehalogenation. The preferential coordination of metavanadate species to high-valence Pd at the carbon-metal interface helps to activate H2 heterolytically for the selective hydrogenation of nitro-groups and inhibits the accumulation of hydroxylamine intermediates. With the mechanism insights, the selective hydrogenation of a range of halogenated nitroaromatics has been successfully achieved by the co-modified Pd catalyst developed in this work.
4-Cl-NB (AR), 2-chloronitrobenzene (AR), 3‑chloronitrobenzene (AR), 3, 4‑dichloronitrobenzene (AR), 1, 2, 3-trichloro-5-nitrobenzene (AR), 4-nitro-1, 2, 3-trichloro-benzene (AR), 2-chloro-5-nitrobenzotrifluoride (AR), 3-chloro-4-fluoro-1-nitrobenzene (AR), 1-fluoro-4-nitrobenzene (AR), 1-fluoro-3-nitrobenzene (AR), 1‑bromo‑4‑nitrobenzene (AR), 1‑bromo‑3‑nitrobenzene (AR), 4-iodonitrobenzene (AR), and benzaldehyde (AR) were purchased from Alfa Aesar Chemical Reagent Co., Ltd. (Tianjin, China). Pd/C catalyst, PPh3 (AR), NaVO3 (AR), and ethanol (AR) were purchased from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Carbon black (Cabot vulcan XC-72) was purchased from Acmec Biochemical Co., Ltd. (Shanghai, China). H2 (99.999%) and D2 (99.999%) were purchased from Linde Gas. The water employed in all experiments was of an ultrapure quality (18.25 MΩ·cm). All reagents were used without any further purification process.
The transmission electron microscopy (TEM), scanning transmission electron microscopy (STEM), and energy dispersive spectroscopy (EDS) elemental mapping measurements were performed on a JEOL JEM-F200 transmission electron microscope. The resolution of TEM and STEM was 0.10 and 0.14 nm at an operating voltage of 200 kV, respectively. The energy resolution of EDS was no greater than 133 eV (Mn Kα). The samples were prepared by dropping ethanol dispersion of samples onto 300-mesh carbon-coated copper grids and immediately evaporating the solvent. The oxygen-isolated 1H and 13C NMR spectra were recorded on Bruker Avance NEO 500 MHz digital FT-NMR spectrometer with tetramethylsilane or solvent signal as an internal reference. All NMR data were processed using MestReNova software. For oxygen-isolated operation, the nuclear magnetic tube was sealed with a silicone rubber gasket and a sealing film, 300 μL of CD3OD was added, and the air inside the tube was replaced with N2 (10 mL·min-1) for 5 min. Aliquots of 200 μL were taken using a 1 mL syringe with a 20 cm needle (φ=0.3 mm). Ethanol was used to fill the syringe needle and keep it free of air. The sample was rapidly taken out and injected into the sealed tube. The tube was purged with N2 for another 5 min and then allowed to stand for 1 h to allow the solid catalysts to settle naturally. The power X-ray diffraction (XRD) experiments were conducted on Rigaku Ultima Ⅳ X-ray diffractometer using Cu Kα radiation (λ=0.154 18 nm) with the operation voltage and current of 40 kV and 30 mA respectively and the scanning range of 5° to 90°. The X-ray photoelectron spectroscopy (XPS) measurements were carried out on an Escalab 250 Xi+ XPS photoelectron spectrometer (Al Kα radiation with 1 486.6 eV operating at 15 kV, 35 W, and 200 μm spot size). The in‑situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) was carried out on a Nicolet iS50 Fourier Transform Infrared spectrometer equipped with a mercury cadmium telluride detector (spectral range: 350-7 800 cm-1). Inductively coupled plasma-optical emission spectrometry (ICP-OES) measurements were carried out on Spectro Spectroblue Fmx36 (wavelength range: 165-770 nm, optical resolution: < 0.008 nm at 200 nm).
Pretreatment of the Pd/C catalyst: 1 g of Pd/C catalyst powder was placed in a 100 mL serum bottle, which was then dried in an oven at a temperature of 60 ℃ for 24 h. Following this, 30.0 mg of the dried Pd/C catalyst (1.41×10-2 mmol) was added into a 10 mL centrifuge tube. The catalyst powder was then dispersed by sonication with the addition of 6 mL of ethanol for further characterization and catalytic performance evaluations. To investigate the influence of oxidation or reduction on the catalytic performance of Pd/C catalysts, 1 g of Pd/C catalyst was placed in an alumina crucible and subjected to the desired temperature and atmosphere (either O2 or H₂), with a heating rate of 2 ℃·min-1 and an oxidation or reduction time of 2 h. Thereafter, the sample was cooled naturally to room temperature for catalysis evaluations. The Pd/C catalyst reduced in a H2 atmosphere at 300 ℃ was denoted as Pd/C-300H. The Pd/C catalyst oxidized in an O2 atmosphere at 300 ℃ was denoted as Pd/C-300O.
Modification of the Pd/C catalyst with PPh3 (denoted as P-Pd/C): 30.0 mg of dried Pd/C catalyst powder was dispersed in 15 mL of ethanol in a 50 mL serum vial. The dispersion was sonicated until the catalyst powder was fully dispersed in ethanol. 18.5 mg of PPh3 (7.05×10-2 mmol) was then added to the mixture. After 30 min stirring, the supernatant was removed by centrifugation. The precipitate was re-dispersed in 6 mL of ethanol for further characterization and catalytic performance evaluations.
Modification of the Pd/C catalyst with PPh3 and NaVO3 (denoted as PV-Pd/C): 30.0 mg of dried Pd/C catalyst powder was placed in a 50 mL serum vial. After 10 mL of ethanol was added, the catalyst mixture was sonicated to achieve a homogeneous suspension. 5 mL of NaVO3 (7.05×10-2 mmol, 8.60 mg) aqueous solution and 18.5 mg of PPh3 (7.05×10-2 mmol) were added to the catalyst mixture. After stirring for 30 min, the supernatant was removed by centrifugation. The precipitate was re-dispersed in 6 mL of ethanol for further characterization and catalytic performance evaluations.
Modification of the Pd/C catalyst with NaVO3 (denoted as V-Pd/C): 30.0 mg of dried Pd/C catalyst powder was placed in a 50 mL serum vial. After 10 mL of ethanol was added to the vial, the mixture was subjected to sonication to achieve a homogeneous dispersion. 5 mL of NaVO3 (7.05×10-2 mmol, 8.60 mg) aqueous solution was then added and stirred for 30 min. Thereafter, the supernatant was removed by centrifugation. The precipitate was collected and re-dispersed in 6 mL of ethanol for further characterization and catalytic performance evaluations.
Synthesis of Pd NPs: the synthesis followed the method developed by Xie et al[24]. 5.0 mg of palladium acetylacetonate was dissolved in the mixture of 0.2 mL of formaldehyde and 10 mL of butylamine to form a homogeneous solution. The solution was then transferred to a 25 mL Teflon-lined autoclave and maintained at 200 ℃ for 3 h. The autoclave was then allowed to cool naturally to room temperature. The product was separated by centrifugation.
Modification of Pd NPs, XC-72, Pd/C-300H, and Pd/C-300O with PPh3 and NaVO3 (denoted as PV-Pd NPs, PV-XC-72, PV-Pd/C-300H, and PV-Pd/C-300O, respectively): the procedure is the same as for modification of the Pd/C catalyst with PPh3 and NaVO3, except that Pd NPs, XC-72, Pd/C-300H, and Pd/C-300O catalysts were used.
A glass pressure vessel (ca. 48 mL) was used as a reactor. Firstly, 1 mL of the Pd/C ethanol dispersion (2.35×10-3 mmol Pd) was pipetted into the vial. 4 mL of ethanol and 157.6 mg of 4-Cl-NB (1 mmol) were then added. The vessel was placed in a water bath at 30 ℃ and stirred thoroughly until the 4-Cl-NB was completely dissolved. The system was then purged with H2 for 2 min to remove O2. The pressure was set to be 1.0×105 Pa of H2 (the starting point of the reaction). ca. 100 μL of reaction mixtures was periodically taken from the reactor to identify the conversion and selectivity by oxygen-isolated NMR[2]. Similar procedures were applied to the catalytic hydrogenation of 4-Cl-NB by P-Pd/C, V-Pd/C, PV-Pd/C, PV-Pd NPs, PV-XC-72, PV-Pd/C-300H, and PV-Pd/C-300O catalysts. To ascertain the kinetic isotope effect (KIE), H2 and D2 were employed as the reactants in the catalytic process with all other experimental parameters remaining consistent. In the catalytic hydrogenation of different halogenated nitroaromatics, 1 mmol of the respective substrates was employed in the catalytic process while all other experimental parameters remained unchanged. To assess the stability of the catalysts, the reaction time was set at 50 min. Following the initial catalytic reaction, 1 mmol of 4-Cl-NB was added to the catalyst for hydrogenation. The reaction was then repeated three times. In the catalytic hydrogenation of benzaldehyde, PV-Pd/C or Pd/C was employed as the catalyst, with 1 mmol of benzaldehyde serving as the substrate. All other experimental parameters were identical.
The primitive catalyst employed in this work was a heterogeneous Pd/C catalyst with a Pd loading (mass fraction) of 5%. TEM and STEM images revealed that the Pd NPs with an average size of ca. 2.6 nm were distributed uniformly on the carbon supports (Fig. 2a, 2b, and S1, Supporting information). The Pd NPs exhibited a uniform particle size distribution, with no large NPs present. Furthermore, STEM-EDS elemental mapping confirmed the well-dispersed nature of Pd NPs on the carbon supports (Fig. 2c-2e). The high-resolution TEM (HRTEM) image of an individual NP revealed lattice fringes with an interplanar spacing of 0.225 nm, corresponding to the (111) plane of Pd (Fig. 2f).

The selective hydrogenation of 4-Cl-NB was first applied to assess the catalytic performance of Pd/C catalysts. The conversion and selectivity of the reaction were determined by oxygen-isolated 1H NMR[2]. As evidenced by the 1H NMR results, the Pd/C catalyst enabled the expeditious 4-Cl-NB hydrogenation, achieving a conversion of > 99.9% within 20 min. However, during the conversion of 4-Cl-NB, the accumulation of 4-chlorophenylhydroxylamine (4-Cl-PHA) was evident (with a percentage of up to 30%), and a considerable amount of dechlorinated product of aniline (AN) was present. With the reaction time extended, 4-Cl-PHA and 4-chloroaniline (4-Cl-AN) were fully converted to the by-product AN due to the hydrogenation-induced dichlorination process (Fig. 3a and S2). To prevent the dechlorination, the organic ligand PPh3 was employed to modify the Pd/C catalyst. As shown in Fig. 3b and S3, the by-product of AN was undetectable throughout the P-Pd/C-catalyzed hydrogenation of 4-Cl-NB, indicating that the coordination of PPh3 on the Pd surface impeded the flat-lying adsorption of the substrate and thereby prevented the dechlorination[6, 25]. However, the conversion rate of 4-Cl-NB was also markedly diminished, with a conversion of less than 20% even after 180 min. It is noteworthy that a significant hydroxylamine accumulation was observed, with the presence of over 80%. This is highly detrimental to the high-purity 4-Cl-AN and poses a significant hazard, as hydroxylamine is explosive[22]. To reduce the 4-Cl-PHA accumulation, vanadium species (NaVO3) were introduced into the catalytic system. The results of the 1H and 13C NMR demonstrated that 4-Cl-NB underwent a relatively rapid reaction to 4-Cl-AN, with a complete conversion time of ca.70 min. In addition, the accumulation of 4-Cl-PHA and the generation of AN were barely observed (Fig. 3c-3e and S4, S5). Thus, the PV-Pd/C catalyst successfully achieved the efficient and selective hydrogenation of 4-Cl-NB to 4-Cl-AN (Table S1). To substantiate the synergistic effect of PPh3 and NaVO3, NaVO3 was introduced into the catalytic system independently. As shown in Fig. 3f and S6, the V-Pd/C catalyst enabled the rapid hydrogenation of 4-Cl-NB without the 4-Cl-PHA accumulation. However, the reaction was accompanied by severe dechlorination. 4-Cl-AN was rarely obtained in the final stage. These results indicate that the co-modification of Pd/C catalysts with PPh3 and NaVO3 is crucial in achieving efficient catalytic hydrogenation of 4-Cl-NB. Furthermore, no obvious decay in the conversion and selectivity was observed after the catalyst was recycled five times, indicating the robustness of the PV-Pd/C catalyst (Fig.S7). TEM images of the catalyst revealed that no notable microstructural alterations were observed after the catalytic reaction, also confirming the high stability of the catalyst (Fig.S8).
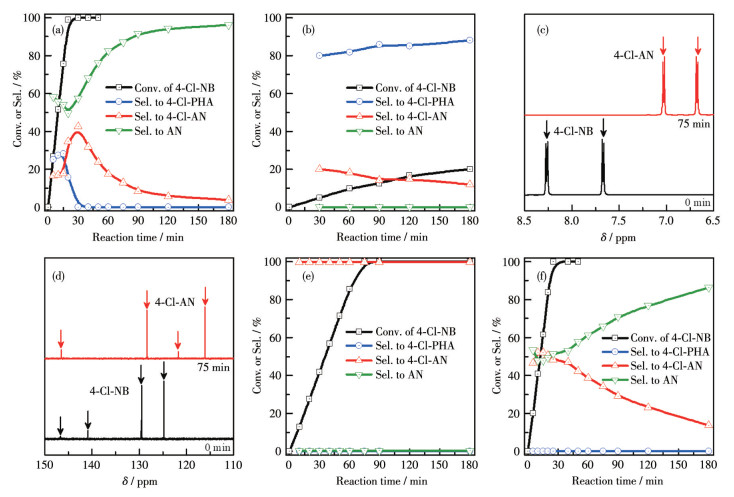
Conv.: conversion; Sel.: selectivity.
To gain insight into how the co-modification of NaVO3 and PPh3 enhanced the catalytic performance of the Pd/C catalysts, we initially conducted a performance comparison to identify the active site. Pd NPs and XC-72 carbon supports were used as the catalysts for comparison. The synthesis of Pd NPs with an average size of ca. 5 nm (Fig.S9) is described in detail in the Experimental methods. As shown in Fig. 4a, compared to the PV-Pd/C catalyst, the PV-Pd NPs and PV-XC-72 displayed almost no catalytic activity. The findings suggest that the co-modification of pure metal surfaces or support surfaces by PPh3 and NaVO3 does not result in the formation of active sites. The metal‑support interface should play a crucial role in the Pd/C catalyst.
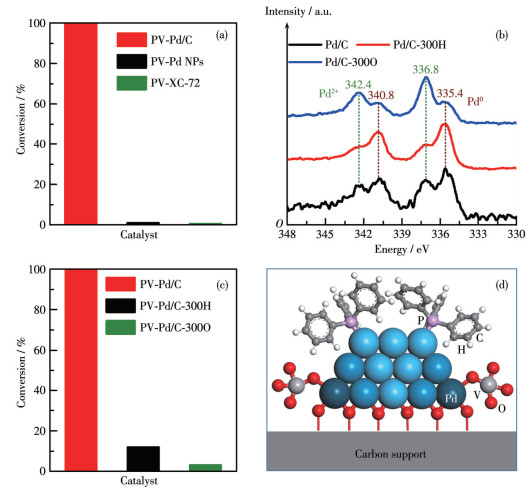
The vanadate species is simply defined as the single V-center structure for clarity in d.
In general, the Pd species at the metal-support interface are observed to be in a high-valence state[26-28]. XPS was employed to ascertain the valence state of the Pd in the Pd/C catalyst. The results revealed that the Pd/C catalyst contained both high- and zero-valence Pd (Fig. 4b, black line). Similarly, the XPS spectra of PV-Pd/C demonstrated that a considerable portion of high-valent Pd species remained although some high-valent Pd species were converted to zero-valence (Fig.S10). To determine the key role of high-valence Pd species at the interface, high-temperature reduction was applied to eliminate as much of most of the high-valence Pd species as possible. When the Pd/C catalyst was reduced in a H2 atmosphere at 300 ℃, the XPS results showed that Pd/C-300H had a greater amount of zero-valence Pd and a significantly reduced amount of high-valence Pd (Fig. 4b, red line), suggesting a reduced amount of high-valence Pd species at the metal-support interface. Moreover, the PV-Pd/C-300H exhibited significantly lower catalytic activity in the catalytic hydrogenation of 4-Cl-NB in comparison to the PV-Pd/C catalyst (Fig. 4c). This result suggests that the high‑valence Pd species at the interface play a key role in creating active sites. After oxidizing the Pd/C catalyst at a high temperature (300 ℃) in an O2 environment, the presence of a considerable amount of high-valence Pd (Fig. 4b, blue line) was revealed by the XPS results. However, the PV-Pd/C-300O resulted in a markedly reduced catalytic activity (Fig. 4c). This result suggests that the presence of high-valence Pd species alone is not enough to optimize the catalytic performance, and further reinforce the need of the simultaneous presence of interfacial high-valence Pd species and surface zero-valence Pd species for enhancing hydrogenation. In addition, the average particle size of Pd NPs in the Pd/C, Pd/C-300H, and Pd/C-300O catalysts was approximately 2.6, 3.0, and 2.3 nm, respectively, suggesting that the small change in the particle size is unlikely to be the primary determinant of the large difference in their catalytic performance (Fig.S1d and S11). The loading and detachment of two modifiers were thus determined by ICP-OES (Table S2 and S3) to evaluate the influence of the quantity of immobilized and free modifiers (Fig.S12). The catalytic performance was not significantly affected by extra modifiers. However, the introduction of enough PPh3 and NaVO3 modifiers was essential to maintain good catalytic performance.
The low catalytic activity of P-Pd/C indicates that PPh3 poisoned the Pd surface. While the PV-Pd/C exhibited superior catalytic activity compared to the P-Pd/C, the PV-Pd NPs showed negligible catalytic activity. These observations suggest that NaVO3 might partially replace the weakly coordinated PPh3. Given the pro-oxidant nature of V and the absence of AN by-product, it is unlikely that NaVO3 would replace the PPh3 on the surface of Pd NPs. The hydrogenation active site of the PV-Pd/C catalyst should not be located at the zero‑valence Pd surface, but rather at the metal‑support interface[29-32]. A schematic structure of the PV-Pd/C catalyst is illustrated in Fig. 4d. The Pd NPs loaded on the carbon support contain both high- and zero-valence Pd species. While the high-valence Pd species are located at the metal-support interface, the zero‑valence Pd species are situated at the metal surface. The PPh3 ligand is coordinated to the zero-valence Pd metal surface via Pd—P bonds. The metavanadate species should be coordinated with the high-valence Pd species at the metal-support interface. Given that the form of vanadate depends on concentration, pH, and the interaction between vanadate and catalyst[33-34], the chemical composition of metavanadate at the interface may be V4O124- or V3O93-.
The in-situ D2 DRIFTS was applied to demonstrate how the PV-Pd/C catalyst activated D2. The generation of substantial O—D species (Fig. 5a) reveals that D2 was activated in a heterolytic manner, resulting in the production of active species of O—D and Pd—D. To ascertain how O—D and Pd—D react with 4-Cl-NB, the KIE was determined (Fig. 5b and S13). The measured KIE value was approximately 2.47, which is a typical secondary KIE and suggests that the rate- determining step of the reaction is the breaking of the chemical bonds of O—H or O—D[14, 35]. These findings indicate that O—D and Pd—D are active species in hydrogenation and that the active sites are located at the metal-support interface rather than the metal surface. If the metal surface is the site of hydrogenation, the measured KIE should be of the first order with 1 < KIE < 2[36]. Given the effectiveness of the O—H and Pd—H in hydrogenating polar C=O unsaturated bonds, the selective hydrogenation of benzaldehyde was employed to further validate that H2 was activated in a heterolytic manner at the interface[26, 28, 35]. As expected, PV-Pd/C selectively converted benzaldehyde to benzyl alcohol with a complete conversion time of ca. 150 min (Fig. 5c). In stark contrast, while the Pd/C catalyst enabled the complete hydrogenation of benzaldehyde within ca. 50 min, the selectivity for benzyl alcohol was negligible (Fig.S14).
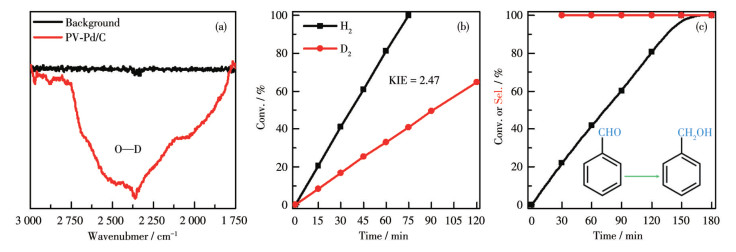
Inset: Conversion of benzaldehyde to benzyl alcohol.
Considering the gained insights, the efficacy of PV-Pd/C in catalyzing the selective hydrogenation of other halogenated nitroaromatics was also evaluated. As shown in Table 1 and Fig.S15, the selective hydrogenation of mono- and polychlorinated nitroaromatics to the corresponding aromatic amines was effectively accomplished by using PV-Pd/C (Entries 1-5), without the influence of other functional groups (i.e., —CF3, —F) (Entries 6-7). Fluorinated, brominated, and iodinated nitroaromatics were also hydrogenated smoothly into the corresponding aromatic amines over the PV-Pd/C catalyst (Entries 8-12).
 下载:
导出CSV
下载:
导出CSV
| Entry | Substrate | Product | Conversion/% | Selectivity/% |
| 1 |  |
 |
ca. 100 | ca. 100 |
| 2 |  |
 |
ca. 100 | ca. 100 |
| 3 |  |
 |
ca. 100 | ca. 100 |
| 4 |  |
 |
ca. 100 | ca. 98 |
| 5 |  |
 |
ca. 100 | ca. 100 |
| 6 |  |
 |
ca. 100 | ca. 100 |
| 7 |  |
 |
ca. 100 | ca. 100 |
| 8 |  |
 |
ca. 100 | ca. 100 |
| 9 |  |
 |
ca. 100 | ca. 99 |
| 10 |  |
 |
ca. 100 | ca. 100 |
| 11 |  |
 |
ca. 100 | ca. 100 |
| 12 |  |
 |
ca. 100 | ca. 99 |
This work demonstrates that the co-modification of Pd/C catalysts with PPh3 and NaVO3 helps to create a high-performance catalyst system for achieving the selective hydrogenation of halogenated nitroaromatics. A systematic investigation revealed that the Pd NPs loaded on the carbon support contain Pd species of mixed valence, with high-valence Pd species at the metal-support interface and zero-valence Pd species on the metal surface. While the strong coordination of PPh3 on the Pd surface helps to inhibit the flat-lying adsorption of halogenated nitroaromatics and thus the dehalogenation process, the preferential coordination of metavanadate on the high-valence Pd species at the metal-support interface enables the efficient conversion of hydroxylamines to amines. Such interfacial active sites facilitate the heterolytic activation of H2 to generate Pd—H and O—H active species. The breakage of O—H bonds is identified as the rate‑determining step in the hydrogenation process. Based on the mechanism understanding, the developed co-modified catalyst is applicable for the selective hydrogenation of a wide range of halogenated nitroaromatics.
CORMA A, SERNA P, CONCEPCIóN P, CALVINO J J. Transforming nonselective into chemoselective metal catalysts for the hydrogenation of substituted nitroaromatics[J]. J. Am. Chem. Soc., 2008, 130(27): 8748-8753. doi: 10.1021/ja800959g
WU Q Y, SU W, HUANG R, SHEN H, QIAO M F, QIN R X, ZHENG N F. Full selectivity control over the catalytic hydrogenation of nitroaromatics into six products[J]. Angew. Chem.‒Int. Edit., 2024, 63(38): e202408731. doi: 10.1002/anie.202408731
SERNA P, CORMA A. Transforming nano metal nonselective particulates into chemoselective catalysts for hydrogenation of substituted nitrobenzenes[J]. ACS Catal., 2015, 5(12): 7114-7121. doi: 10.1021/acscatal.5b01846
SUN X L, WANG Y C, WU Q Y, HAN Y Z, GONG X K, TANG X K, AIKENS C M, SHEN H, ZHENG N F. Cu66 nanoclusters from hierarchical square motifs: Synthesis, assembly, and catalysis[J]. Aggregate, 2024, : e651. doi: 10.1002/agt2.651
BLASER H U, STEINER H, STUDER M. Selective catalytic hydrogenation of functionalized nitroarenes: An update[J]. ChemCatChem, 2009, 1(2): 210-221. doi: 10.1002/cctc.200900129
LI K J, QIN R X, LIU K L, ZHOU W T, LIU N, ZHANG Y Z, LIU S J, CHEN J, FU G, ZHENG N F. Carbon deposition on heterogeneous Pt catalysts promotes the selective hydrogenation of halogenated nitroaromatics[J]. ACS Appl. Mater. Interfaces, 2021, 13(44): 52193-52201. doi: 10.1021/acsami.1c11548
FORMENTI D, FERRETTI F, SCHARNAGL F K, BELLER M. Reduction of nitro compounds using 3d-non-noble metal catalysts[J]. Chem. Rev., 2019, 119(4): 2611-2680. doi: 10.1021/acs.chemrev.8b00547
SHEN H, WU Q Y, MALOLA S, HAN Y Z, XU Z, QIN R X, TANG X K, CHEN Y B, TEO B K, HÄKKINEN H, ZHENG N F. N-heterocyclic carbene-stabilized gold nanoclusters with organometallic motifs for promoting catalysis[J]. J. Am. Chem. Soc., 2022, 144(24): 10844-10853. doi: 10.1021/jacs.2c02669
MITCHELL S, QIN R X, ZHENG N F, PEREZ-RAMIREZ J. Nanoscale engineering of catalytic materials for sustainable technologies[J]. Nat. Nanotechnol., 2021, 16(2): 129-139. doi: 10.1038/s41565-020-00799-8
QIN R X, LIU K L, WU Q Y, ZHENG N F. Surface coordination chemistry of atomically dispersed metal catalysts[J]. Chem. Rev., 2020, 120(21): 11810-11899. doi: 10.1021/acs.chemrev.0c00094
GUO M, LI H, REN Y Q, REN X M, YANG Q H, LI C. Improving catalytic hydrogenation performance of Pd nanoparticles by electronic modulation using phosphine ligands[J]. ACS Catal., 2018, 8(7): 6476-6485. doi: 10.1021/acscatal.8b00872
MAKOSCH M, LIN W I, BUMBáLEK V, Sá J, MEDLIN J W, HUNGERBüHLER K, VAN BOKHOVEN J A. Organic thiol modified Pt/TiO2 catalysts to control chemoselective hydrogenation of substituted nitroarenes[J]. ACS Catal., 2012, 2(10): 2079-2081. doi: 10.1021/cs300378p
ZHAO X J, ZHOU L Y, ZHANG W Y, HU C Y, DAI L, REN L T, WU B H, FU G, ZHENG N F. Thiol treatment creates selective palladium catalysts for semihydrogenation of internal alkynes[J]. Chem, 2018, 4(5): 1080-1091. doi: 10.1016/j.chempr.2018.02.011
WU Q Y, ZHOU W T, SHEN H, QIN R X, HONG Q M, YI X D, ZHENG N F. Surface coordination decouples hydrogenation catalysis on supported metal catalysts[J]. CCS Chem., 2022, 5(5): 1215-1224.
WANG W, XU W L, THAPA K B, YANG X R, LIANG J H, ZHU L Y, ZHU J L. Morpholine-modified Pd/γ-Al2O3@ASMA pellet catalyst with excellent catalytic selectivity in the hydrogenation of p-chloronitrobenzene to p-chloroaniline[J]. Catalysts, 2017, 7(10): 292. doi: 10.3390/catal7100292
MARSHALL S T, O'BRIEN M, OETTER B, CORPUZ A, RICHARDS R M, SCHWARTZ D K, MEDLIN J W. Controlled selectivity for palladium catalysts using self-assembled monolayers[J]. Nat. Mater., 2010, 9(10): 853-858. doi: 10.1038/nmat2849
SCHOENBAUM C A, SCHWARTZ D K, MEDLIN J W. Controlling the surface environment of heterogeneous catalysts using self-assembled monolayers[J]. Acc. Chem. Res., 2014, 47(4): 1438-1445. doi: 10.1021/ar500029y
WU B H, HUANG H Q, YANG J, ZHENG N F, FU G. Selective hydrogenation of α, β-unsaturated aldehydes catalyzed by amine-capped platinum-cobalt nanocrystals[J]. Angew. Chem.‒Int. Edit., 2012, 51(14): 3440-3443. doi: 10.1002/anie.201108593
RUAN P P, CHEN B L, ZHOU Q, ZHANG H S, WANG Y H, LIU K L, ZHOU W T, QIN R X, LIU Z, FU G, ZHENG N F. Upgrading heterogeneous Ni catalysts with thiol modification[J]. The Innovation, 2023, 4(1): 100362. doi: 10.1016/j.xinn.2022.100362
LU L F, ZOU S H, FANG B Z. The critical impacts of ligands on heterogeneous nanocatalysis: A review[J]. ACS Catal., 2021, 11(10): 6020-6058. doi: 10.1021/acscatal.1c00903
CARGNELLO M, CHEN C, DIROLL B T, DOAN-NGUYEN V V, GORTE R J, MURRAY C B. Efficient removal of organic ligands from supported nanocrystals by fast thermal annealing enables catalytic studies on well-defined active phases[J]. J. Am. Chem. Soc., 2015, 137(21): 6906-6911. doi: 10.1021/jacs.5b03333
BAUMEISTER P, BLASER H U, STUDER M. Strong reduction of hydroxylamine accumulation in the catalytic hydrogenation of nitroarenes by vanadium promoters[J]. Catal. Lett., 1997, 49: 219-222. doi: 10.1023/A:1019034128024
STUDER M, NETO S, BLASER H U. Modulating the hydroxylamine accumulation in the hydrogenation of substituted nitroarenes using vanadium-promoted RNi catalysts[J]. Top. Catal., 2000, 13: 205-212. doi: 10.1023/A:1009050804228
ZHANG L, WANG L, JIANG Z Y, XIE Z X. Synthesis of size-controlled monodisperse Pd nanoparticles via a non-aqueous seed-mediated growth[J]. Nanoscale Res. Lett., 2012, 7: 312. doi: 10.1186/1556-276X-7-312
ZHANG J, WANG L, SHAO Y, WANG Y Q, GATES B C, XIAO F S. A Pd@zeolite catalyst for nitroarene hydrogenation with high product selectivity by sterically controlled adsorption in the zeolite micropores[J]. Angew. Chem.‒Int. Edit., 2017, 56(33): 9747-9751. doi: 10.1002/anie.201703938
LIU P X, ZHAO Y, QIN R X, MO S G, CHEN G X, GU L, CHEVRIER D M, ZHANG P, GUO Q, ZANG D D, WU B H, FU G, ZHENG N F. Photochemical route for synthesizing atomically dispersed palladium catalysts[J]. Science, 2016, 352(6287): 797-801. doi: 10.1126/science.aaf5251
SCHALOW T, BRANDT B, STARR D E, LAURIN M, SHAIKHUTDINOV S K, SCHAUERMANN S, LIBUDA J, FREUND H J. Size-dependent oxidation mechanism of supported Pd nanoparticles[J]. Angew. Chem.‒Int. Edit., 2006, 45(22): 3693-3697. doi: 10.1002/anie.200504253
YOU P Y, ZHAN S Q, RUAN P P, QIN R X, MO S G, ZHANG Y Z, LIU K L, ZHENG L S, ZHENG N F. Interfacial oxidized Pd species dominate catalytic hydrogenation of polar unsaturated bonds[J]. Nano Res., 2024, 17: 228-234. doi: 10.1007/s12274-023-5538-9
CHEN G X, ZHAO Y, FU G, DUCHESNE P N, GU L, ZHENG Y P, WENG X F, CHEN M S, ZHANG P, PAO C W, LEE J F, ZHENG N F. Interfacial effects in iron-nickel hydroxide-platinum nanoparticles enhance catalytic oxidation[J]. Science, 2014, 344(6183): 495-499. doi: 10.1126/science.1252553
ZHANG W Y, QIN Q, DAI L, QIN R X, ZHAO X J, CHEN X M, OU D H, CHEN J, CHUONG T T, WU B H, ZHENG N F. Electrochemical reduction of carbon dioxide to methanol on hierarchical Pd/SnO2 nanosheets with abundant Pd-O-Sn interfaces[J]. Angew. Chem.‒Int. Edit., 2018, 57(30): 9475-9479. doi: 10.1002/anie.201804142
WANG Y, QIN R X, WANG Y K, REN J, ZHOU W T, LI L Y, MING J, ZHANG W Y, FU G, ZHENG N F. Chemoselective hydrogenation of nitroaromatics at the nanoscale iron(Ⅲ)-OH-platinum interface[J]. Angew. Chem.‒Int. Edit.., 2020, 59(31): 12736-12740. doi: 10.1002/anie.202003651
吴庆远, 秦瑞轩, 臧丹丹, 张无用, 吴炳辉, 郑南峰. 表面具丰富羟基的介孔TiO2稳定Pt-OH-Fe(Ⅲ)催化界面[J]. 化学学报, 2018,76,(8): 617-621. WU Q Y, QIN R X, ZANG D D, ZHANG W Y, WU B H, ZHENG N F. Stabilizing catalytic Pt-OH-Fe(Ⅲ) interfaces by mesoporous TiO2 with rich surface hydroxyl groups[J]. Acta Chim. Sin., 2018, 76(8): 617-621.
何云峰. 钒(Ⅴ)、(Ⅳ)在水中的形态及钒(Ⅳ)的络合物[J]. 石油与天然气化工, 1983(3): 5-13. HE Y F. Morphology of vanadium(Ⅴ) and (Ⅳ) in water and vanadium(Ⅳ) complexes[J]. Chemical Engineering of Oil & Gas, 1983, (3): 5-13.
SCHOISWOHL J, SURNEV S, NETZER F P, KRESSE G. Vanadium oxide nanostructures: From zero- to three-dimensional[J]. J. Phys.: Condens. Matter, 2006, 18: R1-R14. doi: 10.1088/0953-8984/18/4/R01
WU Q Y, QIN R X, ZHU M S, SHEN H, YU S S, ZHONG Y Y, FU G, YI X D, ZHENG N F. Frustrated Lewis pairs on pentacoordinated Al3+-enriched Al2O3 promote heterolytic hydrogen activation and hydrogenation[J]. Chem. Sci., 2024, 15(9): 3140-3147. doi: 10.1039/D3SC06425E
SHEN H, WU Q Y, HAZER M S, TANG X K, HAN Y Z, QIN R X, MA C X, MALOLA S, TEO B K, HÄKKINEN H, ZHENG N F. Regioselective hydrogenation of alkenes over atomically dispersed Pd sites on NHC-stabilized bimetallic nanoclusters[J]. Chem, 2022, 8(9): 2380-2392. doi: 10.1016/j.chempr.2022.04.017

Figure 1 Reaction network of the hydrogenation of halogenated nitroaromatics, exemplified by 4-chloronitrobenzene (4-Cl-NB)

Figure 2 TEM image (a), STEM image (b), and STEM-EDS elemental mappings (c-e) of Pd/C; (f) HRTEM image of an individual Pd NP

Figure 3 Time-dependent catalysis of 4-Cl-NB hydrogenation on Pd/C (a) and P-Pd/C (b); Oxygen-isolated 1H (c) and 13C (d) NMR spectra for 4-Cl-NB hydrogenation on PV-Pd/C; Time-dependent catalysis of 4-Cl-NB hydrogenation on PV-Pd/C (e) and V-Pd/C (f)
Conv.: conversion; Sel.: selectivity.

Figure 4 (a) Catalytic performances of PV-Pd/C, PV-Pd NPs, and PV-XC-72 in the hydrogenation of 4-Cl-NB (Reaction time: 70 min); (b) XPS spectra of Pd/C, Pd/C-300H, and Pd/C-300O; (c) Catalytic performances of PV-Pd/C, PV-Pd/C-300H, and PV-Pd/C-300O in the hydrogenation of 4-Cl-NB (Reaction time: 70 min); (d) Schematic diagram of PV-Pd/C catalyst
The vanadate species is simply defined as the single V-center structure for clarity in d.

Figure 5 (a) In-situ DRIFTS spectra of D2 activation by PV-Pd/C; (b) KIE of selective 4-Cl-NB hydrogenation by PV-Pd/C; (c) Time-dependent catalysis of benzaldehyde hydrogenation on PV-Pd/C
Inset: Conversion of benzaldehyde to benzyl alcohol.
Table 1. Selective hydrogenation of halogenated nitroaromatics by PV-Pd/C
| Entry | Substrate | Product | Conversion/% | Selectivity/% |
| 1 | |
|
ca. 100 | ca. 100 |
| 2 | |
|
ca. 100 | ca. 100 |
| 3 | |
|
ca. 100 | ca. 100 |
| 4 | |
|
ca. 100 | ca. 98 |
| 5 | |
|
ca. 100 | ca. 100 |
| 6 | |
|
ca. 100 | ca. 100 |
| 7 | |
|
ca. 100 | ca. 100 |
| 8 | |
|
ca. 100 | ca. 100 |
| 9 | |
|
ca. 100 | ca. 99 |
| 10 | |
|
ca. 100 | ca. 100 |
| 11 | |
|
ca. 100 | ca. 100 |
| 12 | |
|
ca. 100 | ca. 99 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: