-
[1]
CAI L X, LI S C, YAN D N, ZHOU L P, GUO F, SUN Q F. Watersoluble redox-active cage hosting polyoxometalates for selective desulfurization catalysis[J]. J. Am. Chem. Soc.,
2018, 140(14):
4869-4876.
doi: 10.1021/jacs.8b00394
-
[2]
HADLINGTON T J, HERMANN M, FRENKING G, JONES C. Low coordinate germanium (Ⅱ) and Tin (Ⅱ) hydride complexes: Efficient catalysts for the hydroboration of carbonyl compounds[J]. J. Am. Chem. Soc.,
2014, 136(8):
3028-3031.
doi: 10.1021/ja5006477
-
[3]
YAO W B, WANG J L, ZHONG A G, WANG S L, SHAO Y L. Transition-metal-free catalytic hydroboration reduction of amides to amines[J]. Org. Chem. Front.,
2020, 7(21):
3515-3520.
doi: 10.1039/D0QO01092H
-
[4]
CHAUDHARI M B, GNANAPRAKASAM B. Recent advances in the metal-catalyzed activation of amide bonds[J]. Chem. Asian J.,
2019, 14(1):
76-93.
doi: 10.1002/asia.201801317
-
[5]
HANADA S, TSUTSUMI E, MOTOYAMA Y, NAGASHIMA H. Practical access to amines by platinum catalyzed reduction of carboxamides with hydrosilanes: Synergy of dual Si-H groups leads to high efficiency and selectivity[J]. J. Am. Chem. Soc.,
2009, 131(41):
15032-15040.
doi: 10.1021/ja9055307
-
[6]
BISAI M K, GOUR K, DAS T, VANKA K, SEN S S. Lithium compound catalyzed deoxygenative hydroboration of primary, secondary and tertiary amides[J]. Dalton Trans.,
2021, 50(7):
2354-2358.
doi: 10.1039/D1DT00364J
-
[7]
SUNADA Y, KAWAKAMI H, IMAOKA T, MOTOYAMA Y, NAGASHIMA H. Hydrosilane reduction of tertiary carboxamides by iron carbonyl catalysts[J]. Angew. Chem.-Int. Edit.,
2009, 48(50):
9511-9514.
doi: 10.1002/anie.200905025
-
[8]
DAS S, ADDIS D, ZHOU S, JUNGE K, BELLER M. Zinccatalyzed reduction of amides: Unprecedented selectivity and functional group tolerance[J]. J. Am. Chem. Soc.,
2010, 132(6):
1770-1771.
doi: 10.1021/ja910083q
-
[9]
CHENG C, BROOKHART M. Iridium-catalyzed reduction of second-ary amides to secondary amines and imines by diethylsilane[J]. J. Am. Chem. Soc.,
2012, 134(28):
11304-11307.
doi: 10.1021/ja304547s
-
[10]
DAS S, WENDT B, MOLLER K, JUNGE K, BELLER M. Two iron catalysts are better than one: A general and convenient reduction of aromatic and aliphatic primary amides[J]. Angew. Chem.-Int. Edit.,
2012, 51(7):
1662-1666.
doi: 10.1002/anie.201108155
-
[11]
LI B, SORTAIS J B, DARCEL C. Unexpected selectivity in rutheniumcatalyzed hydrosilylation of primary amides: Synthesis of secondary amines[J]. Chem. Commun.,
2013, 49(35):
3691-3693.
doi: 10.1039/c3cc39149c
-
[12]
REEVES J T, TAN Z L, MARSINI M A, HAN Z S, XU Y B, REEVES D C, LEE H, LU B Z, SENANAYAKE C H. A practical procedure for reduction of primary, secondary and tertiary amides to amines[J]. Adv. Synth. Catal.,
2013, 355(1):
47-52.
doi: 10.1002/adsc.201200835
-
[13]
BLONDIAUX E, CANTAT T. Efficient metal-free hydrositylation of tertiary, secondary and primary amides to amines[J]. Chem. Commun.,
2014, 50(66):
9349-9352.
doi: 10.1039/C4CC02894E
-
[14]
SIMMONS B J, HOFFMANN M, HWANG J, JACKL M K, GARG N K. Nickel-catalyzed reduction of secondary and tertiary amides[J]. Org Lett.,
2017, 19(7):
1910-1913.
doi: 10.1021/acs.orglett.7b00683
-
[15]
PAN Y X, LUO Z L, XU X, ZHAO H Q, HAN J H, XU L J, FAN Q H, XIAO J L. Ru-catalyzed deoxygenative transfer hydrogenation of amides to amines with formic acid/triethylamine[J]. Adv. Synth. Catal.,
2019, 361(16):
3800-3806.
doi: 10.1002/adsc.201900406
-
[16]
TINNIS F, VOLKOV A, SLAGBRAND T, ADOLFSSON H. Chemoselective reduction of tertiary amides under thermal control: Formation of either aldehydes or amines[J]. Angew. Chem.-Int. Edit.,
2016, 55(14):
4562-4566.
doi: 10.1002/anie.201600097
-
[17]
ZHOU S L, JUNGR K, ADDIS D, DAS S, BELLER M. A convenient and general iron-catalyzed reduction of amides to amines[J]. Angew. Chem.-Int. Edit.,
2009, 48(50):
9507-9510.
doi: 10.1002/anie.200904677
-
[18]
DAS S, JOIN B, JUNGE K, BELLER M. A general and selective copper-catalyzed reduction of secondary amides[J]. Chem. Commun.,
2012, 48(21):
2683-2685.
doi: 10.1039/c2cc17209g
-
[19]
KOVALENKO O O, VOLKOV A, ADOLFSSON H. Mild and selective Et2Zn catalyzed reduction of tertiary amides under hydrosilylation conditions[J]. Org. Lett.,
2015, 17(3):
446-449.
doi: 10.1021/ol503430t
-
[20]
DAS H S, DAS S, DEY K, SINGH B, HARIDASAN R K, DAS A, AHMED J, MANDAL S K. Primary amides to amines or nitriles: A dual role by a single catalyst[J]. Chem. Commun.,
2019, 55(79):
11868-11871.
doi: 10.1039/C9CC05856G
-
[21]
IGARASHI M, FUCHIKAMI T. Transition-metal complex-catalyzed reduction of amides with hydrosilanes: a facile transformation of amides to amines[J]. Tetrahedron Lett.,
2001, 42(10):
1945-1947.
doi: 10.1016/S0040-4039(01)00039-9
-
[22]
DAS S, KARMAKAR H, BHATTACHARJEE J, PANDA T K. Aluminium complex as an efficient catalyst for the chemo-selective reduction of amides to amines[J]. Dalton Trans.,
2019, 48(31):
11978-11984.
doi: 10.1039/C9DT01806A
-
[23]
LEISCHNER T, SUAREZ L A, SPANNENBERG A, JUNGE K, NOVA A, BELLER M. Highly selective hydrogenation of amides catalysed by a molybdenum pincer complex: Scope and mechanism[J]. Chem. Sci.,
2019, 10(45):
10566-10576.
doi: 10.1039/C9SC03453F
-
[24]
ONG D Y, YEN Z H, YOSHII A, IMBERNON J R, TAKITA R, CHIBA S. Controlled reduction of carboxamides to alcohols or amines by zinc hydrides[J]. Angew. Chem.-Int. Edit.,
2019, 58(15):
4992-4997.
doi: 10.1002/anie.201900233
-
[25]
SORRIBES I, LEMOS S C S, MARTIN S, MAYORAL A, LIMA R C, ANDRES J. Palladium doping of towards a general and selective catalytic hydrogenation of amides to amines and alcohols[J]. Catal. Sci. Technol.,
2019, 9(24):
6965-6976.
doi: 10.1039/C9CY02128K
-
[26]
SZOSTAK M, SPAIN M, EBERHART A J, PROCTER D J. Highly chemoselective reduction of amides (primary, secondary, tertiary) to alcohols using SmI2/Amine/H2O under mild conditions[J]. J. Am. Chem. Soc.,
2014, 136(6):
2268-2271.
doi: 10.1021/ja412578t
-
[27]
LAMPLAND N L, HOVEY M, MUKHERJEE D, SADOW A D. Magnesium-catalyzed mild reduction of tertiary and secondary amides to amines[J]. ACS Catal.,
2015, 5(7):
4219-4226.
doi: 10.1021/acscatal.5b01038
-
[28]
YE P Q, SHAO Y L, YE X Z, ZHANG F J, LI R H, SUN J N, XU B H, CHEN J X. Homoleptic bis (trimethylsilyl) amides of yttrium complexes catalyzed hydroboration reduction of amides to amines[J]. Org. Lett.,
2020, 22(4):
1306-1310.
doi: 10.1021/acs.orglett.9b04606
-
[29]
BARGER C J, DICKEN R D, WEIDNER V L, MOTTA A, LOHR T L, MARKS T J. La[N (SiMe3)2]3-catalyzed deoxygenative reduction of amides with pinacolborane: Scope and mechanism[J]. J. Am. Chem. Soc.,
2020, 142(17):
8019-8028.
doi: 10.1021/jacs.0c02446
-
[30]
TAMANG S R, SINGH A, BEDI D, BAZKIAEI A R, WARNER A A, GLOGAU K, McDONALD C, UNRUH D K, FINDLATER M. Polynuclear lanthanide-diketonato clusters for the catalytic hydroboration of carboxamides and esters[J]. Nat. Catal.,
2020, 3(2):
154-162.
doi: 10.1038/s41929-019-0405-5
-
[31]
WENG Z Z, CHEN C L, YE L W, LONG L S, ZHENG L S, KONG X J. Lanthanide-oxo clusters for efficient catalytic reduction of carboxamides[J]. Sci. China-Chem.,
2023, 66(2):
443-448.
doi: 10.1007/s11426-022-1493-y
-
[32]
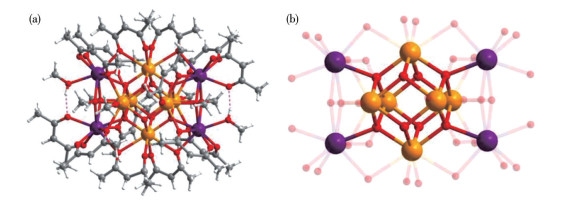
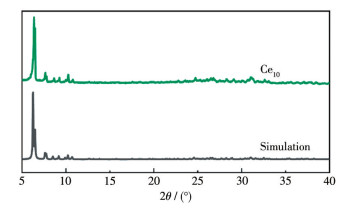
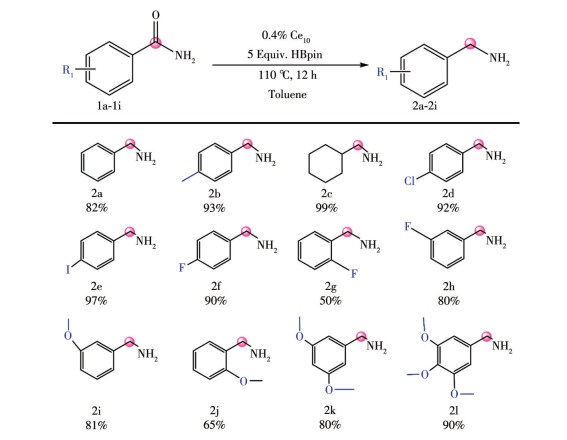
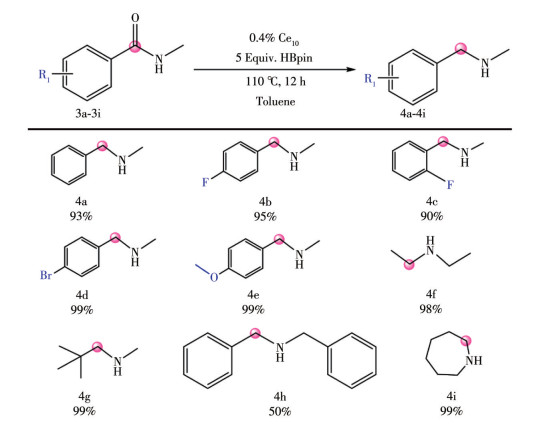
LIU W D, CHEN L Q, QIU Q H, QI M Q, XU H, CHEN C L, LONG L S, ZHENG L S, KONG X J. A mixed valence decanuclear ceriumoxo cluster Ce4ⅢCe6Ⅳ for efficient photocurrent response[J]. Inorg. Chem. Commun.,
2024, 159:
111763.
doi: 10.1016/j.inoche.2023.111763
-
[33]
BAGE A D, HUNT T A, THOMAS S P. Hidden boron catalysis: Nucleophile-promoted decomposition of HBpin[J]. Org. Lett.,
2020, 22(11):
4107-4112.
doi: 10.1021/acs.orglett.0c01168
-
[34]
BROWN H C, HEIM P. Diborane as a mild reducing agent for the conversion of primary, secondary, and tertiary amides into the corresponding amines[J]. J. Am. Chem. Soc.,
1964, 86(17):
3566-3567.
doi: 10.1021/ja01071a037
-
[35]
MULLER F, TRINCADO M, PRIBANIC B, VOGT M, GRUTZMACHER H. Stable BH3 adducts to rhodium amide bonds[J]. J. Organomet. Chem.,
2016, 821:
154-162.
doi: 10.1016/j.jorganchem.2016.05.019
-
[36]
HADEBE S W, ROBINSON R S. Rhodium-catalyzed hydroboration reactions with sulfur and nitrogen analogues of catecholborane[J]. Eur. J. Org. Chem.,
2006, :
4898-4904.
-
[37]
HARDER S, SPIELMANN J. Calcium mediated hydroboration of alkenes: "Trojan horse" or "true" catalysis[J]. J. Organomet. Chem.,
2012, 698:
7-14.
doi: 10.1016/j.jorganchem.2011.09.025
-
[38]
MAITY A, TEETS T S. Main group Lewis acid-mediated transformations of transition-metal hydride complexes[J]. Chem. Rev.,
2016, 116(15):
8873-8911.
doi: 10.1021/acs.chemrev.6b00034
-
[39]
BHUNIA M, SAHOO S R, DAS A, AHMED J, SREEJYOTHI P, MANDAL S K. Transition metal-free catalytic reduction of primary amides using an abnormal NHC based potassium complex: Integrating nucleophilicity with Lewis acidic activation[J]. Chem. Sci.,
2020, 11(7):
1848-1854.
doi: 10.1039/C9SC05953A
-
[40]
YU C, GUO C J, JIANG L H, GONG M L, LUO Y J. Deoxygenation of primary amides to amines with pinacolborane catalyzed by Ca[N (SiMe3)2]2(THF)2[J]. Organometallics,
2021, 40(9):
1201-1206.
doi: 10.1021/acs.organomet.1c00120
-
[41]
ZHANG G Q, WU J, ZHENG S P, NEARY M C, MAO J C, FLORES M, TROVITCH R J, DUB P A. Redox-noninnocent ligand-supported vanadium catalysts for the chemoselective reduction of C=X (X=O, N) functionalities[J]. J. Am. Chem. Soc.,
2019, 141(38):
15230-15239.
doi: 10.1021/jacs.9b07062

 下载:
下载:
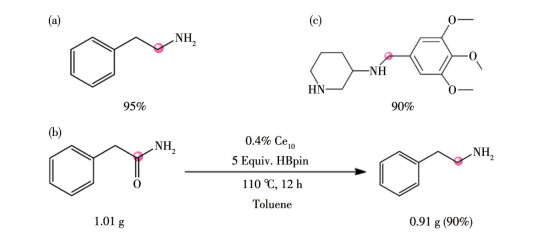
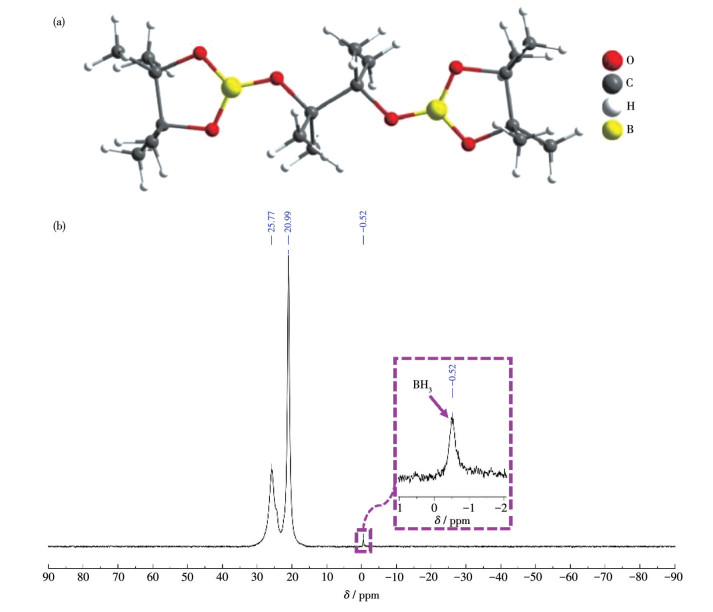
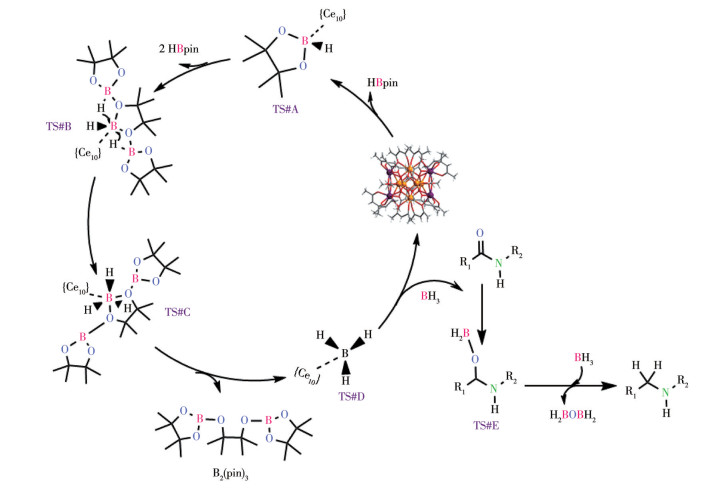

 下载:
下载:








 下载:
下载:
