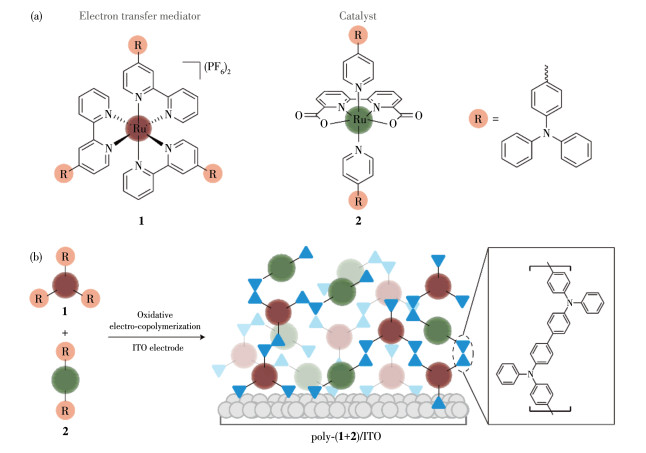
Figure 1.
(a) Molecular structures and (b) schematic representation of the copolymerization of ruthenium complexes 1 and 2
Electro-copolymerized film of ruthenium catalyst and redox mediator for electrocatalytic water oxidation
Hao WANG , Kun TANG , Jiangyang SHAO , Kezhi WANG , Yuwu ZHONG

The construction of a photoelectrochemical or electrolytic cell is a convenient and effective way to split water into H2 and O2 for energy conversion, in which the water oxidation (2H2O→O2+4H++4e-) and reduction (2H++2e-→H2) processes take place on the anodic and cathodic electrode, respectively. The water oxidation process is considered to be the bottleneck of the water splitting reaction due to the high intrinsic redox potential and the high overpotential induced by the accumulation of four electrons. Thus, the development of anodic electrodes with high water-oxidation efficiency is in urgent demand. Modifying electrodes with molecular water oxidation catalysts (WOCs) is a feasible and low-cost method toward this end. Since Meyer and his colleagues conducted a pioneering study in this field[1-2], great efforts have been made to develop highly efficient WOCs and water oxidation systems[3-16]. At present, the ruthenium complex RuⅡ(bda)(L)2 (H2bda is 2, 2′-bipyridine-6, 6′-dicarboxylic acid; L is a neutral donor ligand.) invented by Sun and coworkers exhibits the state-of-the-art performance in water oxidation[5, 17], laying the foundation for the preparation of water-oxidation and splitting devices.
To date, a large number of functional molecular electrodes have been fabricated for water oxidation and splitting based on various film fabrication processes, including chemical adsorption with carboxylate-[18, 19], phosphonate-[20-22], or siloxyl anchoring groups[23-25], layer-by-layer coordination assembly with Zr(Ⅳ)-phosphonate bridges[26-28], and electropolymerization[29-34]. Among these techniques, electropolymerization features easy operation, controllable film coverage and thickness, and rapid electrosynthesis of polymers[35-36]. In particular, research efforts have been made on the reductive electropolymerization of vinyl‑functionalized metal complex catalysts and photosensitizers to prepare functional electrodes for water oxidation[29, 31, 33-34].In contrast, the application of oxidative electropolymerization in this field is rare[30]. Triphenylamine (TPA) is a representative functional group used in the oxidative electropolymerization of metal complexes[37-41]. The resulting complexes are metallopolymers linked with rigid and electron-rich tetraphenylbenzidine bridges. This is in contrast with the metallopolymers obtained via the reductive electropolymerization of vinyl-functionalized metal complexes, which are linked by flexible butane bridges. Considering that the rigid and electron-rich bridges may facilitate the electron transfer among different subcomponents[41], we introduced the TPA units into ruthenium complexes to immobilize complex [Ru(Nbpy)3]2+ (1; with PF6- counteranions; Nbpy=4- triphenylamino-2, 2′-bipyridine) as a redox mediator and complex [Ru(bda)(Npy)2] (2; Npy=4-triphenylaminopyridine) as the WOC by an oxidative electro-copolymerization strategy (Fig. 1). The characterization of these films and the application of the fabricated electrode in the construction of an electrolytic cell for water splitting are presented herein. In addition, the effect of the redox mediator in enhancing electrocatalytic water oxidation is discussed.
Nbpy, [Ru(bda)(DMSO)2], and Npy were synthesized according to the literature method[5, 42-43]. Other reagents were obtained commercially and used without further purification.
To 10 mL ethylene glycol were added RuCl3·3H2O (13 mg, 0.05 mmol) and Nbpy (50 mg, 0.125 mmol). The mixture was heated under microwave conditions (240 W for 3 min, followed by 400 W for 30 min). After cooling to room temperature, an excess of aqueous KPF6 solution was added. The resulting precipitate was collected by filtration and washing with water and Et2O. The obtained orange solid was subjected to flash column chromatography on silica gel (eluent: CH3CN/H2O/aq. KNO3, 200∶10∶1, V/V) to give 25 mg complex 1 in 32% yield. MALDI-TOF: 1 444.2 for [M-PF6]+, 1 298.2 for [M-2PF6]+. 1H NMR (400 MHz, CD3CN): δ 9.21-9.02 (m, 6H), 8.32-8.15 (m, 4H), 8.12 (d, J=5.6 Hz, 1H), 8.09 (d, J=6.0 Hz, 1H), 8.02 (d, J=6.0 Hz, 2H), 7.89 (d, J=6.8 Hz, 6H), 7.87-7.72 (m, 4H), 7.60-7.45 (m, 4H), 7.38 (t, J=8.0 Hz, 12H), 7.17 (t, J=9.6 Hz, 6H), 7.16 (d, J=8.0 Hz, 12H), 7.09 (d, J=8.8 Hz, 6H). Anal. Calcd. for C84H63N9F12P2Ru·H2O(%): C, 62.76; H, 4.08; N, 7.84. Found(%): C, 62.46; H, 4.03; N, 7.94.
To 20 mL MeOH were added [Ru(bda)(DMSO)2] (50 mg, 0.1 mmol) and Npy (64 mg, 0.2 mmol). The mixture was heated at 80 ℃ for 12 h under a N2 atmosphere. The system was cooled to room temperature and the resulting precipitate was collected by filtration. The crude product was purified by silica gel chromatography (eluent: MeOH/CH2Cl2, 1∶100, V/V) to yield 30 mg of complex 2 as a brown powder in 30% yield. MALDI-TOF: 988.1 for [M]+. 1H NMR (300 MHz, DMSO): δ 8.7 (d, J=6 Hz, 2H), 7.96-7.85 (m, 4H), 7.66 (d, J=6 Hz, 4H), 7.60 (d, J=9 Hz, 4H), 7.52 (d, J=6 Hz, 4H), 7.34 (t, J=9 Hz, 8H), 7.14-7.06 (m, 12H), 6.89 (d, J=9 Hz, 4H). Anal. Calcd. for C58H42N6O4Ru·2H2O(%): C, 68.02; H, 4.53; N, 8.21. Found(%): C, 68.32; H, 4.22; N, 8.18.
1H NMR spectra were measured in the designated solvent with a Fourier 300 MHz spectrometer. Mass data were recorded with an Autoflex Ⅲ MALDI-TOF mass spectrometer with α‑cyano‑4‑hydroxycinnamic acid as the matrix. Elemental analysis was performed with Flash EA 1112 or Carlo Erba 1106 analyzer at the Institute of Chemistry, Chinese Academy of Sciences. All electrochemical measurements were carried out in a three-electrode system using a CHI 660D potentiostat. A platinum wire was used as the counter electrode and an Ag/AgCl electrode [197 mV (vs NHE)] as the reference electrode, respectively. The half potential E1/2 was determined by taking the average of the anodic and cathodic peak potentials by E1/2=(Epa+Epc)/2 from the cyclic voltammetry (CV). The gas of hydrogen was detected by gas chromatography-GC7900 (Techcomp), using He as the carrying gas. Scanning electron microscopy (SEM) images were acquired on the Hitachi SU8010 instrument at 10 kV. X-ray photoelectron spectroscopy (XPS) data were collected by the ESCALab220i-XL electron spectrometer from VG Scientific using 300 W Al Kα radiation.
The ITO substrates were used as the working electrode to prepare electropolymerized thin films for electrochemical measurements. The ITO substrates were cleaned by sonicating with detergent water, deionized water, acetone, and ethanol for 15 min, respectively, followed by drying with nitrogen gas. The poly-(1+2)/ITO electrodes were carried out via repeated anodic potential scanning on the cleaned working ITO electrode from 0.0 to 2.0 V at a potential scan rate of 0.1 V·s-1 in 0.1 mol·L-1nBu4NClO4/CH2Cl2 solution containing 0.4 mmol·L-1 of monomer 1 and 0.2 mmol·L-1 of monomer 2. Poly-1/ITO and poly-2/ITO electrodes were obtained by electropolymerization in the corresponding monomer solution with a concentration of 0.2 mmol·L-1. After modification with the electropolymerized thin film, the working electrodes were thoroughly rinsed with CH3CN to remove physically adsorbed materials.
All tests of CV and controlled potential electrolysis were performed on a CHI 660D electrochemical analyzer at room temperature. The measurements were carried out by using the electropolymerized film coated on ITO glass substrate as the working electrode (effective area: 1 cm2), and Ag/AgCl electrode and Pt wire electrode as the reference and counter electrodes, respectively, in 0.1 mol·L-1 aqueous Na2SO4 supporting electrolyte solution unless otherwise stated.
The kinetic isotope effect (KIE) was measured by a chronoamperometry method in 0.1 mol·L-1 aqueous (H2O or D2O) Na2SO4 solution. The charge amounts Q passed through anodes were recorded. The value of KIE (RKIE) was determined by QH/QD, in which QH and QD represent the charge measured in H2O or D2O, respectively.
The oxygen and hydrogen evolution experiments were carried out in an airtight 25 mL three-neck bottle containing 0.1 mol·L-1 Na2SO4 (14 mL) with poly-(1+2)@ITO as the working electrodes, Pt wire as the counter electrode, and Ag/AgCl as the reference electrode, respectively. The gaseous phase (0.1 mL) above the solution was extracted with a syringe (0.1 mL) and injected into GC for analysis.
The CV curves recorded during the electropolymerization process are shown in Fig. 2a. The electro- copolymerized film was obtained on an ITO electrode through the repetitive CV scan mode from a solution consisting of a mixture of complexes 1 (0.4 mmol·L-1) and 2 (0.2 mmol·L-1) in 0.1 mol·L-1nBu4NClO4 electrolyte solution in CH2Cl2, with Pt as the counter electrode and Ag/AgCl as the reference electrode, respectively. When the potential was scanned repeatedly between 0 and 2.0 V at a scan rate of 0.1 V·s-1, the current increased continuously and gradually. After around six cyclic potential scans, the current reached 1.0 mA. The resulting electro‑copolymerized film displayed four consecutive redox peaks at E1/2 of 0.51, 0.83, 1.05, and 1.37 V (vs Ag/AgCl) [0.71, 1.03, 1.25, and 1.57 V (vs NHE)], respectively. The new peaks at 0.83 and 1.05 V could be attributed to the oxidations of the in-situ generated tetraphenylbenzidine segments to aminium radical cations (two N0/N+ processes). The other two peaks at 0.51 and 1.37 V could be assigned to the RuⅡ/RuⅢ processes of complex 2 and 1, respectively. These results indicate that complexes 1 and 2 are successfully co-immobilized on the ITO electrode. The as-prepared electrode was denoted as the poly-(1+2)/ITO electrode, with which all measurements below were carried out unless otherwise stated.
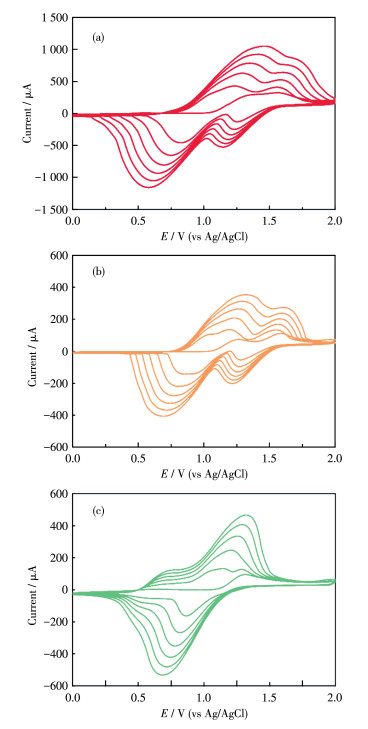
For comparison, the electrodes modified with only complex 1 or complex 2, denoted as poly-1/ITO and poly-2/ITO electrodes, respectively, were prepared by the electropolymerization conducted in the CH2Cl2 solution containing 0.2 mmol·L-1 of 1 or 0.2 mmol·L-1 of 2, respectively (Fig. 2b and 2c). The redox peaks ascribed to the in-situ generated tetraphenylbenzidine segments were observed for both films. However, only one RuⅡ/RuⅢ process was observed at 1.37 V for poly-1 and 0.51 V for poly-2, respectively, due to the oxidation of the corresponding ruthenium component.
XPS and imaging analysis were performed on the obtained films (Fig.S1, Supporting information). These data confirm that these Ru complexes are successfully assembled on the ITO surface by electropolymerization, and the functional molecules are uniformly distributed on the surface. SEM images of the cross sections of these films showed that the thicknesses of these films were 70-80 nm and the surfaces were characterized by domains with sizes of smaller than 500 nm (Fig.S2). It should be noted that the thickness of these films cannot be precisely controlled and we provide in this work only some qualitative analysis of the catalytic activity of films with different compositions.
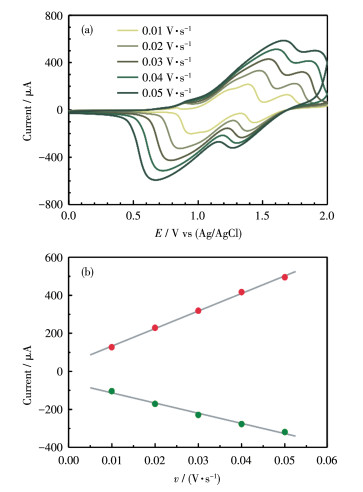
Fig. 3 shows the CVs of poly-(1+2)/ITO film in a clean electrolyte solution at different scan rates (from 0.01 to 0.05 V·s-1). The redox waves of RuⅡ/RuⅢ and N0/N+ processes were retained. Based on the analysis of the RuⅡ/RuⅢ couple at 1.37 V, both the anodic peak current (ipa) and the cathodic peak current (ipc) were linearly dependent on the potential scan rate, indicating that the involved redox reactions are surface‑ controlled rather than diffusion-controlled processes. The surface concentrations (Γ) of complex 1 and catalyst 2 on this poly‑(1+2)/ITO electrode were calculated by Γ=Q/(nFA) to be 6.6×10-9 and 8.3×10-10 mol·cm-2, respectively, in which Q is the integrated charge of the involved peak, F is the Faraday′s constant, and A is the surface area of the electrode, respectively. This corresponds to a molar ratio (n1/n2) of around 8.0 of complex 1 and catalyst 2 on the electrode surface. The relatively low coverage of catalyst 2 on this electrode compared to complex 1 is mainly caused by two factors. Firstly, the concentration of 2 (0.2 mmol·L-1) in the mother solution used in electropolymerization is lower than 1 (0.4 mmol·L-1). Secondly, complex 1 possesses three TPA units, while catalyst 2 only contains two TPA units, leading to a higher electropolymerization efficiency of 1. This electrode is proven to yield an optimal performance of electrocatalytic water oxidation, as discussed below.
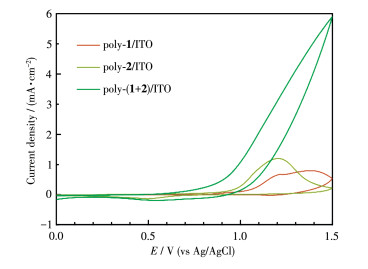
To probe the potential of electrocatalytic water oxidation of these films, the electrodes of poly-1/ITO, poly-2/ITO, and poly-(1+2)/ITO were subjected to CV measurements in Na2SO4 aqueous solutions with pH 6.4 (Fig. 4). The poly-(1+2)/ITO electrode displays a RuⅡ/RuⅢ peak of at 0.52 V (vs Ag/AgCl)[0.72 V (vs NHE)] and an onset potential (Eonset) for water oxidation around 0.87 V (vs Ag/AgCl)[1.07 V (vs NHE)]. The onset overpotential (η) was determined to be 220 mV based on the theoretical potential in pH 6.4. When the potential was scanned over 1.0 V, the current increased significantly due to the efficient water oxidation. In contrast, no remarkable catalytic water oxidation peaks were observed on poly-1/ITO and poly-2/ITO electrodes. This suggests that the coexistence of complexes 1 and 2 is beneficial for water oxidation. The presence of 1 is believed to act as an electron transfer mediator to effectively reduce the catalytic overpotential and thus improve the ability of water oxidation[44].
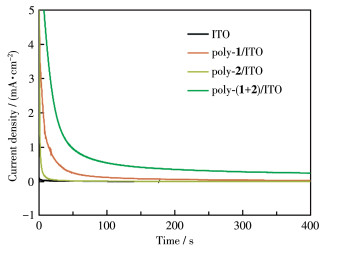
The effectiveness of the poly-(1+2)/ITO electrode for water oxidation was further investigated by controlled potential electrolysis (CPE). An electrolytic cell was constructed with the poly-(1+2)/ITO as the anode (working electrode) in 0.1 mol·L-1 Na2SO4 aqueous solution. A platinum wire and Ag/AgCl electrode were used as the counter and reference electrode, respectively. After electrolysis for 300 s at 1.7 V (vs NHE), the poly-(1+2)/ITO electrode still yielded a catalytic current density of 270 μA·cm-2 (Fig. 5). Small bubbles were observed at the working electrode and counter electrode during the electrolysis process, suggestive of the generation of O2 and H2, respectively, by water splitting. When the CPE measurements were performed at lower applied voltages, the catalytic current density decreased distinctly (Fig.S3). In addition, no catalytic current and bubble evolution were detected for bare ITO, poly-1/ITO, and poly-2/ITO electrodes (Fig. 5). These results further demonstrate that the presence of the electron transfer mediator is critical for the water oxidation electrocatalysis.
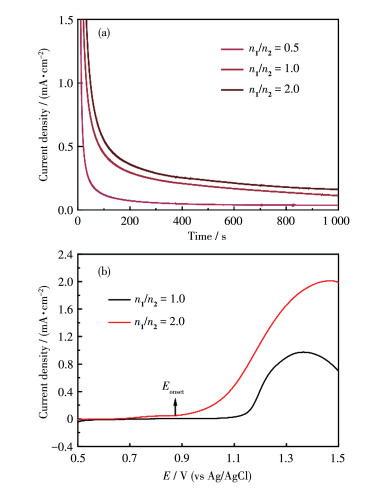
It should be noted that the monomer ratio of complexes 1 and 2 in the mother solution used for the electropolymerization has an important impact on the catalytic performance of the resulting poly-(1+2)/ITO electrode. Specifically, when the electrode was prepared from the solution with a low monomer molar ratio of 0.5 of complex 1 versus catalyst 2, the electrolytic cell showed a low level of catalytic current as demonstrated by the CPE measurement at 1.7 V (vs NHE) (Fig. 6a). When this monomer ratio was increased to 1.0 or 2.0, the obtained electrode showed a significant enhancement of the catalytic current density (other conditions remain unchanged). In addition, the large monomer ratio was beneficial for decreasing Eonset, as demonstrated by the linear sweep voltammetry (LSV) analysis (Fig. 6b). The copolymer electrode prepared with a monomer ratio of 2.0 showed an optimal Eonset of around 0.87 V (vs Ag/AgCl) [1.07 V (vs NHE)], which is significantly lower with respect to the electrode with an equivalent monomer ratio. These results again support that the presence of an excess of the redox mediator of complex 1 is beneficial for improving the catalytic performance.
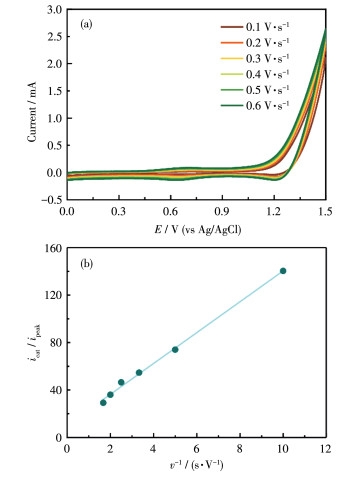
The rate constant of water oxidation (kobs) was examined in 0.1 mol·L-1 Na2SO4 by CV measurements at different scan rates (v) from 0.1 to 0.6 V·s-1 based on the equation icat/ipeak=[4RTncat/(np2F)]kobs/v, where icat is the catalytic current at 1.7 V (vs NHE) corresponding to the water oxidation reaction with four electrons involved (ncat=4), ipeak is the peak current of the RuⅡ/RuⅢ wave with one electron transferred in the redox process (np=1), R is the ideal gas constant, T is the temperature (298 K), respectively[32, 45-46]. Based on the linear relationship of icat/ipeak versus v-1 shown in Fig. 7b, the kobs value for the electrocatalytic water oxidation of poly-(1+2)/ITO was calculated to be about 31.7 s-1. For comparison, a previously reported copolymer film prepared by the reductive electropolymerization of vinyl-functionalized ruthenium complexes presented kobs of 0.073 s-1 at 1.7 V (vs NHE) in an aqueous solution containing 0.1 mol·L-1 NaOAc/HOAc and 0.5 mol·L-1 NaClO4 (pH=4.7)[32]. Though the comparison of different systems should be taken with care, these results do suggest that the poly-(1+2)/ITO film represents an efficient material for electrocatalytic water oxidation.
The gas generated in the electrolytic cell during the CPE measurement by poly-(1+2)/ITO at 1.7 V in 0.1 mol·L-1 aqueous Na2SO4 solution (pH 6.4) was collected and quantified by gas chromatography (GC) analysis. After electrolysis for 1 000 s, a total of 0.32C of charge was passed through the electrode and the generation of H2 was calculated to be about 1.54 μmol (Fig.S4). Accordingly, the Faraday efficiency for H2 generation was calculated to be 93%. Unfortunately, the generated O2 could not be quantified by GC analysis, due to the small amount of O2 and the dissolution of the generated O2 in the electrolyte solution. The initial turnover frequency (TOF) of the poly-(1+2)/ITO electrode for water oxidation under 650 mV overpotential was estimated to be 1.01 s-1 by TOF=Q/(4tFΓ), where Q is the charge passed through the electrode, Γ is the surface coverage of catalyst 2, and t is the electrolysis time, respectively[30].
Considering that Ru(bpy)3 (bpy=2, 2′-bipyridine) derivatives are often used as photosensitizers in photocatalytic reactions[47], we investigated the potential photocatalytic performance of the poly‑(1+2)/ITO electrode. Unfortunately, no gas evolution was detected at an applied bias of up to 0.6 V (vs NHE) in 0.1 mol·L-1 aqueous Na2SO4 solution under visible light irradiation, suggesting that the poly-(1+2)/ITO electrode is not suitable for photocatalysis. This is a common issue for electropolymerized films, which is likely due to the presence of multiple charge traps in these films that can quench the excited states photosensitizers[28-33].
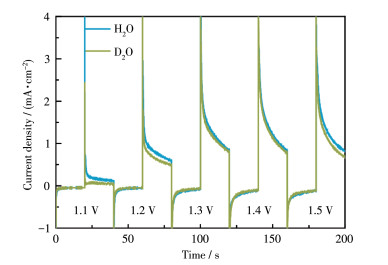
The catalytic mechanism of the RuⅡ(bda)(L)2 catalyst and its derivatives have been explored in depth in literature[5, 17].The formation of the O—O bond, which is the rate-determining step of the O2 evolution, can be achieved through the interaction of two metal-oxo (M=O) units (I2M pathway) or via the water nucleophilic attack to the high valent M=O species (WNA pathway). The water oxidation efficiency based on the I2M mechanism is typically much higher than that of the WNA mechanism[47-48]. This is supported by the fact that some dinuclear ruthenium catalysts show higher water oxidation efficiencies than mononuclear ruthenium catalysts with related structures[49-52]. The catalytic mechanism and efficiency are heavily influenced by the arrangement and distance between the Ru centers of the catalyst. To probe the catalytic mechanism of the poly‑(1+2)/ITO electrode, the KIE was investigated[31, 45]. As shown in Fig. 8, the isotope experiment of poly-(1+2)/ITO was conducted in H2O and D2O solution, respectively, with 0.1 mol·L-1 Na2SO4 as the supporting electrolyte. The RKIE was estimated to be 1.1, suggesting that the O—O bond formation is achieved through I2M pathway (Fig.S5)[5, 17, 31, 40, 45]. The electropolymerized assembly may provide a suitable environment for the approaching of two Ru=O units, favoring the I2M pathway.
An oxidative copolymerization method is used for the preparation of copolymer film electrodes containing the Ru(bpy)3 derivative 1 and WOC 2. The electrolytic cell constructed with this electrode shows an efficient electrocatalytic activity to split water into H2 and O2. An initial TOF of 1.01 s-1 for O2 evolution is achieved in aqueous Na2SO4 solution with pH 6.4 under 650 mV overpotential. Complex 1 is believed to act as an electron transfer mediator to reduce the overpotential of catalysts and improve the water oxidation performance, highlighting the importance of electron transfer mediation in electrocatalytic water oxidation with decreased overpotentials. The rigid electrocopolymer assembly provides an effective method to incorporate different functional groups into the catalytic system, affording a suitable environment for the O—O bond formation via the I2M pathway.
Concepcion J J, Jurss J W, Brennaman M K, Hoertz P G, Patrocinio A O T, Murakami Iha N Y, Templeton J L, Meyer T J. Making oxygen with ruthenium complexes[J]. Acc. Chem. Res., 2009, 42(12): 1954-1965. doi: 10.1021/ar9001526
Gersten S W, Samuels G J, Meyer T J. Catalytic oxidation of water by an oxo-bridged ruthenium dimer[J]. J. Am. Chem. Soc., 1982, 104(14): 4029-4030. doi: 10.1021/ja00378a053
Blakemore J D, Schley N D, Balcells D, Hull J F, Olack G W, Incarvito C D, Eisenstein O, Brudvig G W, Crabtree R H. Half-sandwich iridium complexes for homogeneous water-oxidation catalysis[J]. J. Am. Chem. Soc., 2010, 132(45): 16017-16029. doi: 10.1021/ja104775j
Concepcion J J, Jurss J W, Templeton J L, Meyer T J. One site is enough[J]. catalytic water oxidation by[Ru(tpy)(bpm)(OH2)]2+ and[Ru(tpy)(bpz)(OH2)]2+. J. Am. Chem. Soc., 2008, 130(49): 16462-16463.
Duan L L, Bozoglian F, Mandal S, Stewart B, Privalov T, Llobet A, Sun L C. A molecular ruthenium catalyst with water-oxidation activity comparable to that of photosystem Ⅱ[J]. Nat. Chem., 2012, 4: 418-423. doi: 10.1038/nchem.1301
Kärkäs M D, Åkermark T, Chen H, Sun J L, Åkermark B. A tailor-made molecular ruthenium catalyst for the oxidation of water and its deactivation through poisoning by carbon monoxide[J]. Angew. Chem. Int. Ed., 2013, 52(15): 4189-4193. doi: 10.1002/anie.201210226
Zong R F, Thummel R P. A new family of Ru complexes for water oxidation[J]. J. Am. Chem. Soc., 2005, 127(37): 12802-12803. doi: 10.1021/ja054791m
Wasylenko D J, Ganesamoorthy C, Henderson M A, Koivisto B D, Osthoff H D, Berlinguette C P. Electronic modification of the[RuⅡ(tpy)(bpy)(OH2)]2+ scaffold: Effects on catalytic water oxidation[J]. J. Am. Chem. Soc., 2010, 132(45): 16094-16106. doi: 10.1021/ja106108y
Matheu R, Ertem M Z, Pipelier M, Lebreton J, Dubreuil D, Benet- Buchholz J, Sala X, Tessier A, Llobet A. The role of seven-coordination in Ru-catalyzed water oxidation[J]. ACS Catal., 2018, 8(3): 2039-2048. doi: 10.1021/acscatal.7b03638
Sun K, Saadi F H, Lichterman M F, Hale W G, Wang H P, Zhou X H, Plymale N T, Omelchenko S T, He Z H, Papadantonakis K M, Brunschwig B S, Lewis N S. Stable solar-driven oxidation of water by semiconducting photoanodes protected by transparent catalytic nickel oxide films[J]. Proc. Natl. Acad. Sci. U. S. A., 2015, 112(12): 3612-3617. doi: 10.1073/pnas.1423034112
Zeng P, Zhou Y, Peng L L, Wang S C, Peng T Y. Architecture modification and In3+-doping of WO3 photoanodes to boost the photoelectrochemical water oxidation performance[J]. Sci. China Chem., 2023, 66(11): 3269-3279. doi: 10.1007/s11426-023-1691-1
张永政, 郭旭, 宋欣月, 李欣. 过渡金属电解水催化剂的非金属掺杂研究进展[J]. 无机化学学报, 2024,40,(2): 289-306. ZHANG Y Z, GUO X, SONG X Y, LI X. Advances in non-metallic doping of transition metal electrocatalysts for overall water splitting[J]. Chinese J. Inorg. Chem., 2024, 40(2): 289-306.
樊萌萌, 文晓江, 陶紫阳, 赵强, 李晋平, 刘光. BiVO4/ZnFe2O4同型异质结光阳极的构筑及其光电催化分解水性能[J]. 无机化学学报, 2023,39,(1): 23-31. FAN M M, WEN X J, TAO Z Y, ZHAO Q, LI J P, LIU G. Construction and photoelectrochemical water oxidation performance of BiVO4/ZnFe2O4 homotypic heterojunction photoanode[J]. Chinese J. Inorg. Chem., 2023, 39(1): 23-31.
Qin L S, Zhang W, Cao R. Hydrophilic MnO2 nanowires coating with o-fluoroaniline for electrocatalytic water oxidation[J]. Chin. J. Struct. Chem., 2023, 42(8): 100105. doi: 10.1016/j.cjsc.2023.100105
Fan X Y, Yuan X J, Zhang K. Boosting bulk charge transport of CuWO4 photoanodes via Cs doping for solar water oxidation[J]. Chin. J. Struct. Chem., 2024, 43(2): 100207. doi: 10.1016/j.cjsc.2023.100207
Zhang Y, Wang B, Hu C, Humayun M, Huang Y P, Cao Y L. Negem M, Ding Y G, Wang C D[J]. Fe—Ni—F electrocatalyst for enhancing reaction kinetics of water oxidation. Chin. J. Struct. Chem., 2024, 43(2): 100243.
Wang L, Duan L L, Wang Y, Ahlquist M S G, Sun L C. Highly efficient and robust molecular water oxidation catalysts based on ruthenium complexes[J]. Chem. Commun., 2014, 50(85): 12947-12950. doi: 10.1039/C4CC05069J
Joya K S, Subbaiyan N K, D'Souza F, de Groot H J M. Surface-immobilized single-site iridium complexes for electrocatalytic water splitting[J]. Angew. Chem. Int. Ed., 2012, 51(38): 9601-9605. doi: 10.1002/anie.201203560
Chen Z F, Concepcion J J, Jurs J W, Meyer T J. Single-site, catalytic water oxidation on oxide surfaces[J]. J. Am. Chem. Soc., 2009, 131(43): 15580-15581. doi: 10.1021/ja906391w
Youngblood W J, Lee S H A, Kobayashi Y, Hernandez-Pagan E A, Hoertz P G, Moore T A, Moore A L, Gust D, Mallouk T E. Photoassisted overall water splitting in a visible light-absorbing dye- sensitized photoelectrochemical cell[J]. J. Am. Chem. Soc., 2009, 131(3): 926-927. doi: 10.1021/ja809108y
Zhao Y X, Swierk J R, Megiatto J D, Sherman B, Youngblood W J, Qin D D, Lentz D M, Moore A L, Moore T A, Gust D, Mallouk T E. Improving the efficiency of water splitting in dye-sensitized solar cells by using a biomimetic electron transfer mediator[J]. Proc. Natl. Acad. Sci. U. S. A., 2012, 109(39): 15612-15616. doi: 10.1073/pnas.1118339109
Gao Y, Ding X, Liu J H, Wang L, Lu Z K, Li L, Sun L C. Visible light driven water splitting in a molecular device with unprecedentedly high photocurrent density[J]. J. Am. Chem. Soc., 2013, 135(11): 4219-4222. doi: 10.1021/ja400402d
Wu L, Nayakb A, Shao J, Meyerb T J. Crossing the bridge from molecular catalysis to a heterogenous electrode in electrocatalytic water oxidation[J]. Proc. Natl. Acad. Sci. U. S. A., 2019, 116(23): 11153-11158. doi: 10.1073/pnas.1902455116
Wu L, Eberhart M, Shan B, Nayak A, Brennaman M K, Miller A J M, Shao J, Meyer T J. Stable molecular surface modification of nanostructured, mesoporous metal oxide photoanodes by silane and click chemistry[J]. ACS Appl. Mater. Interfaces, 2019, 11(4): 4560-4567. doi: 10.1021/acsami.8b17824
Alibabaei L, Brennaman M K, Norris M R, Kalanyan B, Song W, Losego M D, Concepcion J J, Binstead R A, Parsons G N, Meyer T J. Solar water splitting in a molecular photoelectrochemical cell[J]. Proc. Natl. Acad. Sci. U. S. A., 2013, 110(50): 20008-20013. doi: 10.1073/pnas.1319628110
Hanson K, Torelli D A, Vannucci A K, Brennaman M K, Luo H, Alibabaei L, Song W, Ashford D L, Norris M R, Glasson C R K, Concepcion J J, Meyer T J. Self-assemble bilayer films of ruthenium(Ⅱ)/polypyridyl complexes through layer-by-layer deposition on nanostructured metal oxides[J]. Angew. Chem. Int. Ed., 2012, 51(51): 12782-12785. doi: 10.1002/anie.201206882
Ding X, Gao Y, Zhang L L, Yu Z, Liu J H, Sun L C. Visible light-driven water splitting in photoelectrochemical cells with supramolecular catalysts on photoanodes[J]. ACS Catal., 2014, 4(7): 2347-2350. doi: 10.1021/cs500518k
Ashford D L, Sherman B D, Binstead R A, Templeton J L, Meyer T J. Electro-assembly of a chromophore-catalyst bilayer for water oxidation and photocatalytic water splitting[J]. Angew. Chem. Int. Ed., 2015, 54(16): 4778-4781. doi: 10.1002/anie.201410944
Wang L, Fan K, Daniel Q, Duan L L, Li F S, Philippe B, Rensmo H, Chen H, Sun J L, Sun L C. Electrochemical driven water oxidation by molecular catalysts in situ polymerized on the surface of graphite carbon electrode[J]. Chem. Commun., 2015, 51(37): 7883-7886. doi: 10.1039/C5CC00242G
Li F S, Fan K, Wang L, Daniel Q, Duan L L, Sun L C. Immobilizing Ru(bda) catalyst on a photoanode via electrochemical polymerization for light-driven water splitting[J]. ACS Catal., 2015, 5(6): 3786-3790. doi: 10.1021/cs502115f
Wu L, Brennaman M K, Nayak A, Eberhart M, Miller A J M, Meyer T J. Stabilization of ruthenium(Ⅱ) polypyridyl chromophores on mesoporous TiO2 electrodes: Surface reductive electropolymerization and silane chemistry[J]. ACS Central Sci., 2019, 5(3): 506-514.
Ashford D L, Lapides A M, Vannucci A K, Hanson K, Torelli D A, Harrison D P, Templeton J L, Meyer T J. Water oxidation by an electropolymerized catalyst on derivatized mesoporous metal oxide electrodes[J]. J. Am. Chem. Soc., 2014, 136(18): 6578-6581.
Sherman B D, Ashford D L, Lapides A M, Sheridan M V, Wee K R, Meyer T J. Light-driven water splitting with a molecular electroassembly-based core/shell photoanode[J]. J. Phys. Chem. Lett., 2015, 6(16): 3213-3217.
Zhong Y W, Yao C J, Nie H J. Electropolymerized films of vinyl- substituted polypyridine complexes: Synthesis, characterization, and applications[J]. Coord. Chem. Rev., 2013, 257(7/8): 1357-1372.
Gu C, Huang N, Gao J, Xu F, Xu Y H, Jiang D L. Controlled synthesis of conjugated microporous polymer films: Versatile platforms for highly sensitive and label-free chemo-and biosensing[J]. Angew. Chem. Int. Ed., 2014, 53(19): 4850-4855.
Li M, Li Y F. Solid-phase electrosynthesis[J]. Acc. Chem. Res., 2023, 56(24): 3694-3703.
Shao J Y, Zhong Y W. Stabilization of a cyclometalated ruthenium sensitizer on nanocrystalline TiO2 by an electrodeposited covalent layer[J]. Inorg. Chem., 2019, 58(5): 3509-3517.
Cui B B, Mao Z P, Chen Y X, Zhong Y W, Yu G, Zhan C L, Yao J N. Tuning of resistive memory switching in electropolymerized metallopolymeric films[J]. Chem. Sci., 2015, 6(2): 1308-1315.
Wang J X, Zhang H, Li S M, Ding C J, Zhao Y J, Long X Z, Wei C, Wang Y F, Li Y F, Shen L Y, Cui S X, Hong W J, Li M. Crystalline unipolymer monolayer with high modulus and conductivity[J]. Angew. Chem. Int. Ed., 2022, 62(4): e202216838.
Zhang J, Du J, Wang J X, Wang Y F, Wei C, Li M. Vertical step-growth polymerization driven by electrochemical stimuli from an electrode[J]. Angew. Chem. Int. Ed., 2018, 57(51): 16698-16702.
Li P, Wang Z X, Song C P, Zhang H Y. Rigid fused p-spacers in D-π-A type molecules for dye-sensitized solar cells: A computational investigation[J]. J. Mater. Chem. C, 2017, 5(44): 11454.
Zhao J, Dang F F, Liu B A, Wu Y, Yang X L, Zhou G J, Wu Z X, Wong W Y. Bis-ZnⅡ salphen complexes bearing pyridyl functionalized ligands for efficient organic light-emitting diodes (OLEDs)[J]. Dalton Trans., 2017, 46(18): 6098-6110.
Vannucci A K, Alibabaei L, Losego M D, Concepcion J J, Kalanyan B, Parsons G N, Meyer T J. Crossing the divide between homogeneous and heterogeneous catalysis in water oxidation[J]. Proc. Natl. Acad. Sci. U. S. A., 2013, 110(52): 20918-20922.
Sheridan M V, Sherman B D, Fang Z, Wee K R, Coggins M K, Meyer T J. Electron transfer mediator effects in the oxidative activation of a ruthenium dicarboxylate water oxidation catalyst[J]. ACS Catal., 2015, 5(7): 4404-4409.
Vannucci A K, Hull J F, Chen Z F, Binstead R A, Concepcion J J, Meyer T J. Water oxidation intermediates applied to catalysis: Benzyl alcohol oxidation[J]. J. Am. Chem. Soc., 2012, 134(9): 3972-3975.
Walden A G, Miller A J M. Rapid water oxidation electrocatalysis by a ruthenium complex of the tripodal ligand tris(2-pyridyl)phosphine oxide[J]. Chem. Sci., 2015, 6(4): 2405-2410.
Li A, Zhang Y F, Sun Z C, Niu Z Y, Lan G X. Photosensitizing metal-organic layers for photocatalysis, artificial photosynthesis and fluorescence imaging[J]. Sci. China Chem., 2023, 66(12): 3372-3382.
Romain S, Vigara L, Llobet A. Oxygen-oxygen bond formation pathways promoted by ruthenium complexes[J]. Acc. Chem. Res., 2009, 42(12): 1944-1953.
Duan L L, Tong L P, Xu Y H, Sun L C. Visible light-driven water oxidation: From molecular catalysts to photoelectrochemical cells[J]. Energy Environ. Sci., 2011, 4(9): 3296-3313.
Jiang Y, Li F, Zhang B B, Li X N, Wang X H, Huang F, Sun L C. Promoting the activity of catalysts for the oxidation of water with bridged dinuclear ruthenium complexes[J]. Angew. Chem. Int. Ed., 2013, 25(12): 3398-3401.
Zhang L L, Gao Y, Ding X, Yu Z, Sun L C. High-performance photoelectrochemical cells based on a binuclear ruthenium catalyst for visible-light-driven water oxidation[J]. ChemSusChem, 2014, 7(10): 2801-2804.
Duan L L, Fischer A, Xu Y H, Sun L C. Isolated seven-coordinate Ru(Ⅳ) dimer complex with[HOHOH]- bridging ligand as an intermediate for catalytic water oxidation[J]. J. Am. Chem. Soc., 2009, 131(30): 10397-10399.

Figure 1 (a) Molecular structures and (b) schematic representation of the copolymerization of ruthenium complexes 1 and 2

Figure 2 CV curves recorded during the electropolymerization process on ITO surface in 0.1 mol•L-1 nBu4NClO4/CH2Cl2 solution containing (a) a mixture of complex 1 (0.4 mmol•L-1) and complex 2 (0.2 mmol•L-1), (b) 0.2 mmol•L-1 of 1, and (c) 0.2 mmol•L-1 of 2 to give poly-(1+2)/ITO, poly-1/ITO, and poly-2/ITO film, respectively

Figure 3 (a) CV curves of poly-(1+2)/ITO in 0.1 mol•L-1 nBu4NClO4/CH2Cl2 at varying scan rates of 0.01, 0.02, 0.03, 0.04, and 0.05 V•s-1; (b) Linear dependence of peak current of the RuⅡ/RuⅢ wave at 1.2 V as a function of scan rate

Figure 4 CV curves of poly-1/ITO, poly-2/ITO, and poly-(1+ 2)/ITO electrodes in Na2SO4 aqueous solution with pH 6.4

Figure 5 CPE with different electrodes (active electrode area: 1 cm2) in 0.1 mol•L-1 Na2SO4 aqueous solution at 1.7 V (vs NHE)

Figure 6 (a) CPE analysis in 0.1 mol•L-1 Na2SO4 aqueous solution at 1.7 V (vs NHE) with different poly-(1+2)/ITO electrodes prepared by the electropolymerization in the mother solution containing different n1/n2 values; (b) LSV curves measured with different poly-(1+2)/ITO electrodes

Figure 7 (a) CV curves of poly-(1+2)/ITO in 0.1 mol•L-1 Na2SO4 solution at varying scan rates from 0.1 to 0.6 V•s-1; (b) Plots of icat/ipeak vs v-1 for poly-(1+2)/ITO

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:

 下载:
下载: