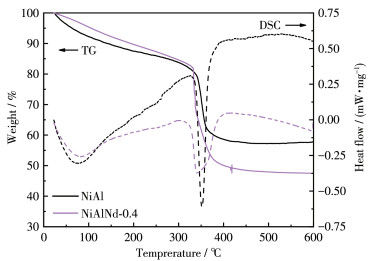
Figure 1.
TG-DSC curves of NiAl and NiAlNd-0.4 catalyst precursors
NiAlNd catalysts for CO2 methanation derived from the layered double hydroxide precursor
Jinglin CHENG , Xiaoming GUO , Tao MENG , Xu HU , Liang LI , Yanzhe WANG , Wenzhu HUANG

The excessive use of fossil fuels leads to a rapid increase in CO2 concentration in the atmosphere and further global warming. CO2 utilization and emissions reduction have become an urgent problem and attracted extensive attention. Although CO2 is the main greenhouse gas, it is also an economical and sustainable carbon source. The strategy of"carbon capture and utilization"(CCU) converting the captured CO2 into the high value - added chemicals or fuels can achieve carbon neutrality in a real sense[1-3].
The CO2 hydrogenation to methane, also called the Sabatier reaction, is one of the most promising technical routes for CO2 conversion and utilization[4-6]. In this route, the hydrogen can be produced by the water electrolysis using the renewable electricity. Because of the"intermittent "nature, renewable energy such as wind, solar, and tidal energies, needs a large-scale energy storage solution[7-8]. With the Sabatier reaction, renewable electricity energy can be stored in methane gas in the form of chemical energy, and this is the so-called power to gas (PtG) concept[9-10].
Ni-based catalysts are widely used for CO2 methanation because of their high catalytic activity and lower cost[4, 11]. Various supports including TiO2[12-13], SiO2[14-15], ZrO2[16-17], Al2O3[18-19] and CeO2[20] have been employed to support the Ni component. However, for Ni-based catalysts, especially those with a high Ni loading, it is usually difficult to achieve well-dispersed Ni particles[21-22]. Layered double hydroxides (LDHs), called hydrotalcite-like materials, consist of layered metal hydroxides and interlayer anions[23]. With high specific surface area and fine - tuning of basicity, the catalysts derived from the LDHs precursors exhibit superior catalytic performances compared with the commercial Ni- based catalyst (usually prepared by the impregnation technique). As a result, these LDHs - derived catalysts have been applied for CO2 methanation in recent years[21, 24].
To further improve the activity of catalysts, several specific methods were developed for catalyst preparation[25-27]. For example, Szabados et al. prepared NiAl4-LDHs by impregnating different Ni - salt solutions over the dry - milled Al(OH)3[25]. Adding appropriate promoters is the most common way for the improvement of catalyst performance[11, 21]. Rare-earth elements can increase the dispersion of Ni particles and the stability of catalysts and regulate the surface acidity/basicity of catalysts. Therefore, some rare-earth elements such as Y[28-29], Sm[30-31], Tb[32], Ce[33-34] and La[35-37] were adopted for the modification of CO2 methanation catalysts. For instance, Sun et al. found that the incorporation of Ce promoted the dispersion of Ni and increased the number of medium basic sites[34]. In addition to modifying the Ni dispersion and surface basicity of the catalyst, the introduction of La improves the redox performance of the catalyst[35]. Neodymium (Nd), an indispensable rare - earth element in the field of magnetic material, has been employed as the promotor for catalysts in recent years. Gac et al. prepared the Nd-promoted Ni/CeO2 and Ni/Al2O3 catalysts for CO2 methanation reaction by the impregnation method, and they found that the introduction of Nd resulted in a great increase in the catalytic activity of Ni/Al2O3 catalyst[38]. As mentioned above, the catalysts based on the LDHs precursor exhibit excellent catalytic properties for CO2 methanation. However, to our knowledge, there is still a lack of investigation concerning the NiAlNd composite oxide catalyst derived from the LDHs precursor.
In this work, a series of NiAlNd catalysts based on LDHs precursor were prepared and used for the CO2 methanation. The main purpose is to investigate the effects of Nd on the physicochemical and catalytic properties of catalysts. Varies of techniques were employed to characterize the catalysts. Moreover, the relationship between the physicochemical properties and catalytic performance was discussed.
All of the chemical reagents (analytical grade) used in the experiment were purchased from Shanghai Titan Technology Co., Ltd., China. The LDHs precursors of NiAlNd catalysts were prepared by the solvent-thermal method. First, nickel nitrate, aluminum nitrate, neodymium nitrate, and urea were dissolved in a mixture of deionized water and glycol under constant stirring at room temperature. The as - formed suspension was transferred to a hydrothermal autoclave and kept at 160 ℃ for 3 h, and the precipitate was collected and washed thoroughly with deionized water and alcohol by turn. Afterward, the product was dried at 80 ℃ overnight to obtain the hydrotalcite precursors. Finally, the precursors were calcined at 400 ℃ for 2 h in static air. To get the LDHs structure, the molar ratio of Ni to Al+ Nd (nNi∶(nAl+nNd)) was kept at a constant of 2.1, which is an optimized value for the CO2 methanation NiAl catalyst[35, 39], and the catalysts were denoted as NiAlNd - x (x represents the molar ratio of Nd to the total amount of Al and Nd (nNd∶(nAl+nNd)), x=0, 0.1, 0.2, 0.3, 0.4, 0.5).
A certain amount of samples were completely dissolved in aqua regia, and the solution was diluted to a suitable concentration using the same nitric acid (2.5 mol·L-1) diluent as the standard solution. The inductively coupled plasma - atomic emission spectrometer (ICP - AES, Thermo IRIS Intrepid IIXSP) was used to quantitatively analyze the concentration of each metal element in the solution, and the content of each metal element in the sample was calculated according to the dilution ratio.
The thermogravimetric -differential scanning calorimeter (TG - DSC) analysis was performed on the STA 449 - F3 thermal analyzer (NETZSCH). About 10 mg samples were used for the test, ranging from 30 to 600 ℃ with a rate of 5 ℃·min-1 in the N2 atmosphere.
The structure analysis of the catalyst was carried out on the X'PERT diffractometer (PANalytical) using Cu Kα rays (λ =0.154 nm) with an operating voltage of 40 kV and an operating current of 35 mA. The scan range was 5°-80° of the scanning speed of 5 (°)·min-1.
The N2 adsorption - desorption test was performed using the ASAP 2020 HD88 instrument (Micromeritics). The sample was first degassed under vacuum at 200 ℃ for 10 h, followed by N2 adsorption and desorption at liquid nitrogen temperature (-196 ℃). The specific surface areas (SBET) of the catalysts were calculated from the linear part of the Brunauer- Emmett-Teller (BET) plot, as well as the pore size and pore volume of the samples were calculated based on the Barrett - Joyner-Halenda model.
Temperature - programmed reduction (TPR) measurements were performed in a linear quartz microreactor (i. d. 4 mm). First, about 50 mg sample was purged with N2 at 300 ℃ for 1 h to remove adsorbed water and other contaminants. After cooling to 50 ℃, the reactor was switched to H2 - N2 mixture gas with a molar ratio of 1∶9 (50 mL·min-1) till the baseline was stable. The temperature was heated to 800 ℃ at 5 ℃· min-1, and the consumption of H2 was measured in the experiment utilizing the thermo-conductivity detector.
Temperature-programmed desorption of CO2 (CO2-TPD) was performed on a self-built device, and the CO2 signal was detected by a mass spectrometer (Pfeiffer Vacuum Quadstar, 32-bit). The 50 mg sample was first reduced at 500 ℃ with an H2 - N2 mixture gas with a molar ratio of 1∶9 for 1 h. Then, the reactor was cooled down to room temperature, and the catalyst was exposed to CO2 for 30 min to reach saturated adsorption. After purging with He for 30 min, CO2 -TPD started by heating the sample to 500 ℃ at 5 ℃·min-1.
X - ray photoelectron spectroscopy (XPS) analysis was performed on an ESCALAB 250Xi spectrometer (Thermo Scientific), using Al Kα as the emission source, and C1s (284.6 eV) was used to correct the binding energy of the measured elements. In the case of the reduced catalyst, the calcined sample was first transferred to a separate reaction chamber under an ultrahigh vacuum by a transfer rod and then exposed to an H2-N2 mixture flow with a molar ratio of 1∶9 (30 mL·min-1) at 500 ℃ for 2 h. After the reduction, the samples were transferred back to the analysis chamber without exposure to air.
The surface area of metallic Ni was measured by the CO chemisorption method. A mass spectrometer (Pefeiffer Vacuum Quadstar, 32-bit) was used to detect the gas signal of CO. First, the 50 mg sample was reduced using H2 - N2 mixture gas with a molar ratio of 1∶9 at 500 ℃ for 2 h, and then cooled to 30 ℃ in Ar atmosphere. The CO/Ar (5∶95, V/V) was injected into the reactor via a six-way valve equipped with a quantitative loop until the catalyst was saturated with CO. The Ni surface area was calculated assuming that the CO adsorption on metallic Ni is a linear adsorption mode and the effective coverage area of a single Ni atom is 1.6×10-19 m2 according to the literature[40].
The evaluation of the catalyst for CO2 hydrogenation to methane was carried out in a continuous flow fixed bed reactor. For each test, 0.15 g catalyst (40-60 mesh) was loaded into the quartz reaction tube, and 0.45 g quartz sand was used to dilute the catalyst to avoid the occurrence of temperature runaway. First, the catalyst was reduced in an H2 - N2 mixture gas with a molar ratio of 1∶9 at 500 ℃ for 4 h. After the reactor cooled down to 150 ℃, the reactor was switched to the feed gas mixture comprising CO2, H2, and N2 in a molar ratio of 18∶72∶10, and then heated to the reaction temperature at a rate of 2 ℃·min-1. The catalytic reaction was carried out at atmospheric pressure, and the space velocity was 24 000 mL·g-1·h-1. The effluent was analyzed online with a gas chromatograph (7820A, Agilent) equipped with a TCD and an FID. The products of CO2 hydrogenation to methane mainly contain CO2, CO, and methane. The CO2 conversion and methane selectivity were calculated as the following formulations[41].
|
|
(1) |
|
|
(2) |
Where XCO2 represents the CO2 conversion; SCH4 is the selectivity of methane; A0x represents the peak area of x component (x means different reactants or products) detected with gas chromatography before the catalytic reaction, and A*x is the peak area of x component after reaction; fx is the correction factor of x component relative to N2. The steady - state values are the average of four analyses taken after 2 h on stream operation.
The precursors of NiAl and NiAlNd - 0.4 catalysts were chosen for the TG - DSC analysis, and the results are presented in Fig. 1. For the NiAl precursor, a weight loss accompanied by an endothermic peak occured in the range of 30-330 ℃ corresponding to the removal of interlayer anion and water. A steep weight loss appeared in the 330 - 400 ℃ range, and a strong endothermic peak was observed at about 350 ℃ simultaneously. The process is related to the decomposition of hydroxide in the LDH precursor. At a temperature higher than 400 ℃, the mass of the catalyst is almost stable, indicating that the precursor had been fully decomposed into oxides. Therefore, a temperature of 400 ℃ was adopted for the calcination of the precursor. The NiAlNd-0.4 sample exhibited similar TG and DSC profiles to the NiAl sample, but the total weight loss of NiAlNd-0.4 was larger than that of the NiAl sample.
The specific surface areas of NiAlNd catalysts were tested by N2 adsorption - desorption, and the results are shown in Table 1. The SBET of the NiAl catalyst is 215 m2·g-1, and the large surface area relates to that the catalyst is derived from a precursor with the LDHs structure. As a small amount of Nd was introduced, no significant change in the SBET could be found. However, when the substitution of Nd3+ for Al3+ reached 0.5, the SBET steeply decreased to 127 m2·g-1. This is because, with the increase in the substitution of Nd3+ for Al3+, the LDHs structure of the catalyst precursor is damaged severely, resulting in a large loss of the SBET. The pore volume (Vpore) and size (Dpore) increased first and then decreased with the increase in the amount of Nd.
 下载:
导出CSV
下载:
导出CSV
| Samples | SBET / (m2·g-1) | Vpore / (cm3·g-1) | Dpore / nm | dNiOa / nm | SNib / (m2·g-1) |
| NiAl | 215 | 0.29 | 5.3 | 3.4 | 23.2 |
| NiAlNd-0.1 | 212 | 0.36 | 6.7 | 3.0 | 24.8 |
| NiAlNd-0.2 | 223 | 0.57 | 10.3 | 2.9 | 27.7 |
| NiAlNd-0.3 | 216 | 0.41 | 7.6 | 2.5 | 21.9 |
| NiAlNd-0.4 | 202 | 0.40 | 8.0 | 2.3 | 21.8 |
| NiAlNd-0.5 | 127 | 0.21 | 6.5 | 1.9 | 10.9 |
| a Determined with Scherrer equation using the diffraction peak at 2θ of 43.4°; b The surface of metallic Ni estimated from CO chemisorption assuming that the ratio of CO to surface Ni atom is 1∶1. | |||||
Fig. 2 shows the SEM images of NiAl and NiAlNd-0.4 catalysts. The sphere - shaped lamellar structure, a typical morphologic feature of LDHs - derived catalyst, was observed for the NiAl sample. The NiAlNd-0.4 catalyst exhibited similar sphere-shaped morphology, however, the radius of the sphere was much smaller than that of NiAl and the lamellar structure feature began to fade. The TEM pictures of NiAl and NiAlNd -0.4 catalysts are shown in Fig. 3. The NiAl sample took on a thin petal -like morphology, and for the NiAlNd-0.4 catalyst, the size of the petal decreased and the feature of the particle can be observed partly. These results indicate that with the introduction of Nd, the lamellar feature of LDHs is destroyed in a certain degree, but the particle size of the catalyst is small.
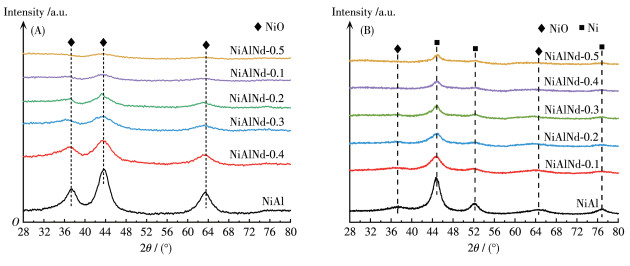
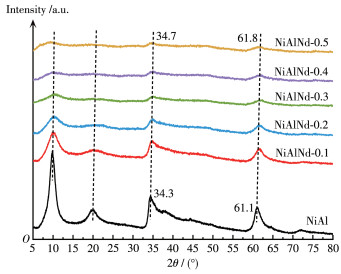
Fig. 4 shows the X - ray diffraction (XRD) patterns of the precursors. The NiAl precursor exhibited a typical LDHs structure, and the diffraction peaks located at 9.9°, 19.9°, 34.3°, and 61.1° correspond to the (003), (006), (009), and (112) planes of LDHs, respectively. With the substitution of Nd3+ for Al3+, the intensities of diffraction peaks reduced markedly suggesting that Nd hinders the formation of LDHs structure. Moreover, the diffraction peaks shift towards a high angle slightly with the increase in Nd amount. This indicates that partial anions such as CO32- and OH- in the interlayer of LDHs move out, decreasing the interlayer distance. The XRD patterns of the calcined catalyst are presented in Fig. 5A. The disappearance of characteristic peaks of LDHs indicates the collapse of LDHs structure. The diffraction peaks at 37.4°, 43.4°, and 63.0° are assigned to the characteristic peak of NiO, and they correspond to the diffraction of (111), (200), and (220) planes, respectively. With the increase in the Nd amount, the diffraction peak intensity of NiO decreased accompanied by an increase in the half - peak width. The grain sizes of NiO (dNiO) in the catalyst are presented in Table 1, and the size decreases from 3.4 to 1.9 nm from NiAl to NiAlNd-0.5. There is no characteristic diffraction peak of Nd2O3 and Al2O3 phases in the pattern, indicating that Nd2O3 and Al2O3 exist in an amorphous state or are dispersed in the catalyst so high as not to be detected. In addition, no diffraction peak of the NiAl2O4 spinel phase was observed in the calcined catalyst, but the existence of NiAl2O4 spinel can't be completely ruled out. Two reasons are responsible for the absence of the diffraction peak of NiAl2O4. On the one hand, the low calcination temperature (400 ℃) leads to the NiAl2O4 phase with a low crystallinity, which can not be detected by the XRD technique[42-43]. On the other hand, the diffraction signal of the NiAl2O4 phase is overlapped with that of the NiO phase[42]. Fig. 5B shows the XRD patterns of the reduced catalyst, and the diffraction peaks appearing at 44.9°, 52.2°, and 76.9° are ascribed to the characteristic peaks of metallic Ni. It is noted that the small diffraction peaks of NiO can still be observed, suggesting that the NiO hasn't been reduced completely at 500 ℃.
For the reduced catalysts, the catalysts were reduced at 500 ℃ for 2 h.
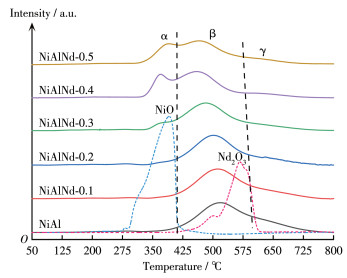
H2 -TPR experiments were conducted on the catalysts, and the results are shown in Fig. 6 For comparison, the TPR profiles of the unitary NiO and Nd2O3 are also presented in Fig. 6 For the Nd - free NiAl sample, the H2 consumption can be divided into two sections. One appears at 400 - 600 ℃ (β section), and the other occurs at 600 - 750 ℃ (γ section). Compared with the TPR profile of the unitary NiO, the reduction of NiO in the NiAl sample shifts towards higher temperatures. The β section can be ascribed to the reduction of NiO strongly interacting with Al2O3[44], and the γ section corresponds to the decrease in NiAl2O4 spinel[45]. With the introduction of Nd, an H2 consumption band appears at 325 -400 ℃ (α section). This is because the addition of Nd weakens the interaction between NiO and Al2O3, and a part of the NiO can break loose from the composite oxides of NiAl and be"free "NiO. For the same reason, the peak temperature in the β section decreases with the increase in the amount of Nd. It should be mentioned that the H2 consumption in the β section includes the reduction of Nd2O3. Compared with the decrease in unitary Nd2O3, the Nd2O3 in the NiAlNd catalyst is reduced at a lower temperature because of its high dispersion. Moreover, in the γ section, the H2 consumption decreases from NiAl to NiAlNd - 0.5, suggesting the introduction of Nd depresses the formation of NiAl2O4 spinel.
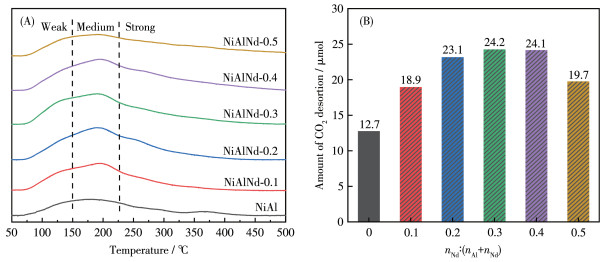
The CO2 - TPD profiles of the catalysts are shown in Fig. 7A. A broad desorption band from 75 to 450 ℃ was observed for all the investigated samples. The desorption band can be roughly divided into three regions: 50 -150 ℃, 150-225 ℃, and > 225 ℃, and they correspond to the CO2 desorption from the weak, medium, and strong basic sites, respectively. The weak basic sites are related to the surface hydroxyl group, i.e. the Brönst alkaline site, over which the bicarbonate (HCO3-) is formed[46-47]. The medium basic site is the oxygen vacancies, and the CO2 molecule can insert into oxygen vacancies and form the bidentate carbonate (b-CO32-)[48-49]. The unsaturated O2- acts as the strong basic site, and the monodentate carbonate (m - CO32-) is formed by the coordination binding of CO2 to unsaturated O2- [48-49]. There was no obvious change in the temperature of the CO2 desorption peak over the NiAl catalysts with/without Nd, indicating that the influence of Nd on the strength of the surface basic site is negligible. The total amounts of CO2 desorption are shown in Fig. 7B. With the increase in Nd amount, the CO2 desorption amount increased and then reached a plateau. However, for the NiAlNd- 0.5, the CO2 desorption amount decreased again, which can be attributed to its smaller specific surface area. The adsorption activation of CO2 occurs at the basic site, and the increase in the number of basic sites is favorable for the increase in the catalytic activity.
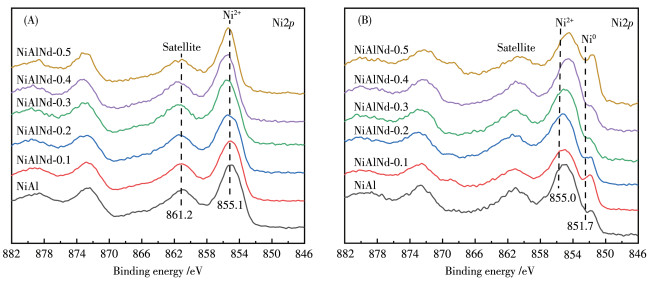
The surface compositions and chemical states of NiAlNd catalysts were studied with XPS technology. As shown in Fig. 8A, for all the calcined samples, the binding energies at 855.1 and 861.2 eV are attributed to the characteristic photoelectron peak of Ni2+2p3/2 and its satellite peak, respectively. After the reduction, a photoelectron peak at 851.7 eV corresponding to the binding energies of Ni02p3/2 was observed for all the catalysts (Fig. 8B). The photoelectron signals of Ni2+2p3/2 still appear because NiO can't be reduced completely at the reduction temperature of 500 ℃, and this is in good accordance to the result of XRD[50]. The surface metal distributions are presented in Table 2. For comparison, the bulk compositions of the catalyst determined by ICP - AES are also listed in Table 2. The atomic-fraction of Ni on the catalyst surface was much lower than that in the bulk phase. However, for the atomic-fraction of Al, a contrary change was observed. In other words, the catalysts demonstrate the surface characteristic of"Al-enrichment and Ni-poor", a general phenomenon for the NiAl composite oxides[35, 51]. Furthermore, the surface Ni atomic- fraction over the NiAlNd (24.3%-21.6%) were higher than that over the NiAl sample (19.9%), suggesting that the substitution of Nd for Al increases the distribution of Ni on the catalyst surface.
下载:
导出CSV
| Sample | Atomic fraction of bulk metala / % | Atomic fraction of surface metalb / % | |||||
| Ni | Al | Nd | Ni | Al | Nd | ||
| NiAl | 70.9(68.0) | 29.1(32.0) | — | 19.9 | 80.1 | 0.0 | |
| NiAlNd-0.1 | 69.2(68.0) | 28.3(28.8) | 2.5(3.2) | 24.3 | 71.6 | 4.1 | |
| NiAlNd-0.2 | 69.1(68.0) | 25.5(25.6) | 5.4(6.4) | 23.4 | 72.2 | 4.4 | |
| NiAlNd-0.3 | 67.2(68.0) | 23.8(22.4) | 9.0(9.6) | 23.2 | 70.9 | 5.9 | |
| NiAlNd-0.4 | 68.2(68.0) | 20.4(19.2) | 11.4(12.8) | 23.7 | 69.0 | 7.2 | |
| NiAlNd-0.5 | 65.7(68.0) | 18.6(16.0) | 15.7(16.0) | 21.6 | 69.3 | 9.1 | |
| a The bulk metal atomic fraction was determined by ICP-AES, and the value in the parenthesis was the nominal value; b The surface metal atomic fraction was determined by XPS. | |||||||
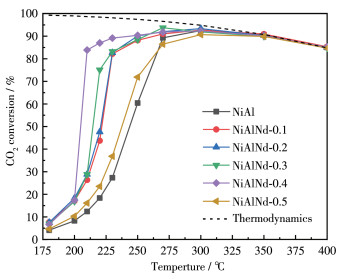
The catalytic activity for CO2 methanation was tested over NiAlNd catalysts in the temperature range of 180-400 ℃, and the results are shown in Fig. 9. The temperature rise is favorable for CO2 conversion from the kinetics perspective but unfavorable in terms of the thermodynamics because the CO2 methanation is an exothermic reaction. In this case, in the temperature range of 180 - 300 ℃, the CO2 conversion increased with the elevation in the reaction temperature, and a jump was observed at 200 - 250 ℃. After reaching 300 ℃, the CO2 conversion began to decrease. The reaction at 180 - 300 ℃ is in the kinetic control zone, while at a temperature higher than 300 ℃, the reaction is limited by thermodynamics. The activity variation is in agreement with the typical behavior of the CO2 methanation. As shown in Fig. 9, in the low-temperature region, the CO2 conversion improves remarkably with the introduction of Nd, and the optimum activity is obtained over the NiAlNd-0.4 catalyst. For example, at 210 ℃, the CO2 conversion over the NiAl catalyst was only 12.4%, and the CO2 conversion on the NiAlNd -0.4 sample was 83.9%, which was almost seven times that of the NiAl catalyst. The CO2 conversion decreased significantly as the molar ratio of Nd to the total amount of Al and Nd reached 0.5. The selectivity of methane was stable and close to 100% at the testing temperature, and only a small amount of CO was produced (less than 0.5%) at temperatures above 350 ℃.
As well known, the adsorption/activation of both H2 and CO2 are involved in the CO2 hydrogenation process. The surface basic site acts as the active site for the adsorption and activation of CO2. The results of CO2 - TPD show that the introduction of Nd increases the number of basic sites, which is also favorable for CO2 adsorption and activation. The metallic Ni is the active site for the adsorption and activation of H 2, and the number and intrinsic activity of the Ni site influence the activities of the catalyst. The specific surface area of metallic Ni (SNi), representing the number of Ni active sites, was measured by the CO chemisorption method, and the results are listed in Table 1. With the introduction of Nd, the SNi increased and a maximum of 27.7 m2·g-1 was obtained on the NiAlNd - 0.2. Further increasing the amount of Nd led to a decrease in the SNi. Especially for the NiAlNd - 0.5 sample, the SNi decreased steeply, and the complete collapse of the LDHs structure of the sample should be responsible for this change. The reducibility of NiO is an important factor affecting the intrinsic activity of the Ni site[35-36]. In general, the better reducibility of the NiO corresponds to the higher intrinsic activity of the Ni[35-36]. As demonstrated in the result of TPR, with the increase in the amount of Nd, the reducibility of the NiO component was enhanced, which means an increase in the intrinsic activity of the Ni site. As the amount of Nd increases from 0 to 0.2, both the number and the intrinsic activity of the Ni site increase, thus, the CO2 conversion increases from NiAl to the NiAlNd - 0.2 sample. For the samples with an amount of Nd higher than 0.2, although the SNi decreased, the intrinsic activity of the Ni site increased. The increase in intrinsic activity prevails over the decrease in the number of Ni sites for the samples of NiAlNd-0.3 and NiAlNd-0.4, while the contrary effect is for the NiAlNd - 0.5 sample. Therefore, the highest CO2 conversion was obtained on NiAlNd-0.4.
In this study, a series of NiAlNd catalysts based on the LDHs precursors were synthesized, and the effects of Nd doping on the NiAl catalyst were investigated. The main conclusions are as follows.
(1) The precursors of NiAlNd catalysts with the LDHs structure can be prepared using the glycol solvent-thermal method, and the LDHs structural characteristics gradually fade away with the increase in the Nd amount.
(2) The introduction of Nd greatly improves the low-temperature activity of the catalyst. Under the condition of 210 ℃, WHSV=24 000 mL·g-1·h-1 and p=100 kPa, the CO2 conversion over the NiAlNd - 0.4 catalyst reaches 83.9%, which is almost seven times that of Nd-free NiAl sample.
(3) Although the Nd doping depresses the formation of the LDHs structure of the precursor, the particle size of the calcined catalyst decreases at the same time. The introduction of Nd weakens the interaction between NiO and Al2O3 and promotes the reduction of NiO and the intrinsic activity of the Ni site. Moreover, the Nd doping increases the amount of base site.
(4) The CO2 methanation activities over NiAlNd catalysts are related to the amount and intrinsic activity of Ni site simultaneously. Furthermore, the increase in the base site is also favorable for the increase in the catalytic activity.
Centi G, Quadrelli E A, Perathoner S. Catalysis for CO2 conversion: A key technology for rapid introduction of renewable energy in the value chain of chemical industries[J]. Energy Environ. Sci., 2013, 6: 1711-1731. doi: 10.1039/c3ee00056g
Zhang X G, Buthiyappan A, Jewaratnam J, Metselaar H S C, Raman A A A. Bifunctional materials for integrated CO2 capture and conver-sion: Review on adsorbent and catalyst types, recent advances, and challenges[J]. J. Environ. Chem. Eng., 2024, 12: 111799. doi: 10.1016/j.jece.2023.111799
Tao Y, Edwards R W J, Mauzerall D L, Celia M A. Strategic carbon dioxide infrastructure to achieve a low-carbon power sector in the mid-western and south-central United States[J]. Environ. Sci. Technol., 2021, 55(22): 15013-15024. doi: 10.1021/acs.est.1c03480
Ashok J, Pati S, Hongmanorom P, Zhang T X, Chen J M, Kawi S. A review of recent catalyst advances in CO2 methanation processes[J]. Catal. Today, 2020, 356: 471-489. doi: 10.1016/j.cattod.2020.07.023
Wang Y, Winter L R, Chen J G, Yan B. CO2 hydrogenation over heterogeneous catalysts at atmospheric pressure: From electronic proper-ties to product selectivity[J]. Green Chem., 2021, 231: 249-267.
Mebrahtu C, Krebs F, Abate S, Perathoner S, Centi G, Palkovits R. CO2 methanation: Principles and challenges[J]. Stud. Surf. Sci. Catal., 2019, 178: 85-103.
Notton G, Nivet M L, Voyant C, Paoli C, Darras C, Motte F, Fouilloy A. Intermittent and stochastic character of renewable energy sources: Consequences, cost of intermittence and benefit of forecasting[J]. Renew. Sustain. Energy Rev., 2018, 87: 96-105. doi: 10.1016/j.rser.2018.02.007
Abujarad S Y, Mustafa M W, Jamian J J. Recent approaches of unit commitment in the presence of intermittent renewable energy resources: A review[J]. Renew. Sustain. Energy Rev., 2017, 70: 215-223. doi: 10.1016/j.rser.2016.11.246
Wulf C, Linßen J, Zapp P. Review of power-to-gas projects in Europe[J]. Energy Procedia, 2018, 155: 367-378. doi: 10.1016/j.egypro.2018.11.041
Ghaib K, Ben-Fares F Z. Power - to - methane: A state - of - the - art review[J]. Renew. Sustain. Energy Rev., 2018, 81: 433-446. doi: 10.1016/j.rser.2017.08.004
Frontera P, Macario A, Ferraro M, Antonucci P. Supported catalysts for CO2 methanation: A review[J]. Catalysts, 2017, 7(2): 59. doi: 10.3390/catal7020059
Zhou R, Rui N, Fan Z, Liu C J. Effect of the structure of Ni/TiO2 catalyst on CO2 methanation[J]. Int. J. Hydrogen Energy, 2016, 41(47): 22017-22025. doi: 10.1016/j.ijhydene.2016.08.093
Le T A, Kim M S, Lee S H, Kim T W, Park E D. CO and CO2 methanation over supported Ni catalysts[J]. Catal. Today, 2017, 293-294: 89-96. doi: 10.1016/j.cattod.2016.12.036
Zhao B R, Liu L, Shi H F, Zhang H G, Zhang J, Wang Y Z, Xie Y X. Plasma - induced micro - combustion for the synthesis of Ni - M/SiO2 (M=La, Ce, Zr) catalysts with high selectivity toward CO2 methana-tion[J]. Ind. Eng. Chem. Res., 2022, 61: 3877-3888. doi: 10.1021/acs.iecr.1c04300
Wang K, Men Y, Liu S, Wang J G, Li Y Y, Tang Y H, Li Z P, An W, Pan X L, Li L. Decoupling the size and support/metal loadings effect of Ni/SiO2 catalysts for CO2 methanation[J]. Fuel, 2021, 304: 121388. doi: 10.1016/j.fuel.2021.121388
Wang H, Li Z Y, Cui G Q, Wei M. Synergistic catalysis at the Ni/ ZrO(2-x) interface toward low - temperature CO2 methanation[J]. ACS Appl. Mater. Interfaces, 2023, 15: 19021-19031. doi: 10.1021/acsami.3c01544
Zhao K C, Wang W H, Li Z H. Highly efficient Ni/ZrO2 catalysts prepared via combustion method for CO2 methanation[J]. J. CO2 Util., 2016, 16: 236-244. doi: 10.1016/j.jcou.2016.07.010
Champon I, Bengaouer A, Chaise A, Thomas S, Roger A C. Carbon dioxide methanation kinetic model on a commercial Ni/Al2O3 catalyst[J]. J. CO2 Util., 2019, 34: 256-265. doi: 10.1016/j.jcou.2019.05.030
Song F J, Zhong Q, Yu Y, Shi M G, Wu Y H, Hu J H, Song Y. Obtaining well-dispersed Ni/Al2O3 catalyst for CO2 methanation with a microwave-assisted method[J]. Int. J. Hydrogen Energy, 2017, 42(7): 4174-4183. doi: 10.1016/j.ijhydene.2016.10.141
Tada S, Shimizu T, Kameyama H, Haneda T, Kikuchi R. Ni/CeO2 catalysts with high CO2 methanation activity and high CH 4 selectivity at low temperatures[J]. Int. J. Hydrogen Energy, 2012, 37(7): 5527-5531. doi: 10.1016/j.ijhydene.2011.12.122
Huynh H L, Yu Z X. CO2 methanation on hydrotalcite-derived catalysts and structured reactors: A review[J]. Energy Technol., 2020, 8(5): 1901475. doi: 10.1002/ente.201901475
He L, Lin Q Q, Liu Y, Huang Y Q. Unique catalysis of Ni -Al hydrotalcite derived catalyst in CO2 methanation: Cooperative effect between Ni nanoparticles and a basic support[J]. J. Energy Chem., 2014, 23(5): 587-592. doi: 10.1016/S2095-4956(14)60144-3
Fan G L, Li F, Evans D G, Duan X. Catalytic applications of layered double hydroxides: Recent advances and perspectives[J]. Chem. Soc. Rev., 2014, 43(20): 7040-7066. doi: 10.1039/C4CS00160E
Abate S, Barbera K, Giglio E, Deorsola F, Bensaid S, Perathoner S, Pirone R, Centi G. Synthesis, characterization, and activity pattern of Ni-Al hydrotalcite catalysts in CO2 methanation[J]. Ind. Eng. Chem. Res., 2016, 55(30): 8299-8308. doi: 10.1021/acs.iecr.6b01581
Szabados M, Szabados T, Mucsi R, Sápi A, Kónya Z, Kukovecz Á, Pálinkó I, Sipos P. Facile preparation of nickel -poor layered double hydroxides from mechanochemically pretreated gibbsite with a variety of interlamellar anions and their use as catalyst precursors for CO2 hydrogenation[J]. Mater. Res. Bull., 2023, 157: 112010. doi: 10.1016/j.materresbull.2022.112010
Namvar F, Salavati-Niasari M, Meshkani F. Effect of the rare earth metals (Tb, Nd, Dy) addition for the modification of nickel catalysts supported on alumina in CO2 methanation[J]. Int. J. Hydrogen Energy, 2023, 48(5): 1877-1891. doi: 10.1016/j.ijhydene.2022.10.096
Xu Y, Chen Y, Li J, Zhou J, Song M, Zhang X Q, Yin Y X. Improved low-temperature activity of Ni-Ce/γ-Al2 O3 catalyst with layer structural precursor prepared by cold plasma for CO2 methanation[J]. Int. J. Hydrogen Energy, 2017, 42(18): 13085-13091. doi: 10.1016/j.ijhydene.2017.04.019
Sun C, Świrk Da Costa K, Wierzbicki D, Motak M, Grzybek T, Da Costa P. On the effect of yttrium promotion on Ni - layered double hydroxides - derived catalysts for hydrogenation of CO2 to methane[J]. Int. J. Hydrogen Energy, 2021, 46(22): 12169-12179. doi: 10.1016/j.ijhydene.2020.03.202
Takano H, Kirihata Y, Izumiya K, Kumagai N, Habazaki H, Hashi-moto K. Highly active Ni/Y - doped ZrO2 catalysts for CO2 methanation[J]. Appl. Surf. Sci., 2016, 388: 653-663. doi: 10.1016/j.apsusc.2015.11.187
Li H, Liu J, Yang J, Ma L Z, Fan X, Liang P, Liu Q, Zhao P W, Wang B, Cheng Y. Why the one-pot synthesized Sm-modified nickel phyllosilicate is more active than the post synthesized one for CO2 methanation? Identifying the pivotal role of generating Sm2Si2O7[J]. Fuel Process.Technol., 2023, 247: 107802. doi: 10.1016/j.fuproc.2023.107802
Liu Q, Yang H Y, Dong H, Zhang W, Bian B, He Q K, Yang J, Meng X B, Tian Z W, Zhao G M. Effects of preparation method and Sm2O3 promoter on CO methanation by a mesoporous NiO-Sm2O3/Al2O3 catalyst[J]. New J. Chem., 2018, 42(15): 13096-13106. doi: 10.1039/C8NJ02282H
Namvar F, Hajizadeh-Oghaz M, Mahdi M A, Ganduh S H, Meshkani F, Salavati-Niasari M. The synthesis and characterization of Ni-M - Tb/Al2O3 (M: Mg, Ca, Sr and Ba) nanocatalysts prepared by different types of doping using the ultrasonic-assisted method to enhance CO2 methanation[J]. Int. J. Hydrogen Energy, 2023, 48(10): 3862-3877. doi: 10.1016/j.ijhydene.2022.10.243
Zhang Q W, Xu R N, Liu N, Dai C N, Yu G Q, Wang N, Chen B H. In situ Ce-doped catalyst derived from NiCeAl-LDHs with enhanced low - temperature performance for CO2 methanation[J]. Appl. Surf. Sci., 2022, 579: 152204. doi: 10.1016/j.apsusc.2021.152204
Sun C, Beaunier P, Da Costa P. Effect of ceria promotion on the catalytic performance of Ni/SBA - 16 catalysts for CO2 methanation[J]. Catal. Sci. Technol., 2020, 10(18): 6330-6341. doi: 10.1039/D0CY00922A
Zhou L L, Guo X M, Hu X, Zhang Y X, Cheng J L, Guo Q S. CO2 methanation reaction over La - modified NiAl catalysts derived from hydrotalcite-like precursors[J]. Fuel, 2024, 362: 130888. doi: 10.1016/j.fuel.2024.130888
Wang Z L, Zhang T Y, Reina T R, Huang L, Xie W F, Musyoka N M, Oboirien B, Wang Q. Enhanced low - temperature CO2 methanation over La - promoted NiMgAl LDH derived catalyst: Fine - tuning La loading for an optimal performance[J]. Fuel, 2024, 366: 131383. doi: 10.1016/j.fuel.2024.131383
Yang H, Wen X Y, Yin S Y, Zhang Y X, Wu C E, Xu L, Qiu J, Hu X, Xu L L, Chen M D. The construction of the Ni/La2O2CO3 nanorods catalysts with enhanced low-temperature CO2 methanation activities[J]. J. Ind. Eng. Chem. Res., 2023, 128: 167-183. doi: 10.1016/j.jiec.2023.07.046
Gac W, Zawadzki W, Kuśmierz M, Słowik G, Grudziński W. Neo-dymium promoted ceria and alumina supported nickel catalysts for CO2 methanation reaction[J]. Appl. Surf. Sci., 2023, 631: 157542. doi: 10.1016/j.apsusc.2023.157542
Dias Y R, Perez-Lopez O W. CO2 methanation over Ni - Al LDH - derived catalyst with variable Ni/Al ratio[J]. J CO2 Util., 2023, 68: 102381. doi: 10.1016/j.jcou.2022.102381
Brooks C S, Kehrer V J. Chemisorption of carbon monoxide on metal surfaces by pulse chromatography[J]. Anal. Chem., 1969, 41: 103-106.
Guo X P, Peng Z J, Hu M X, Zuo C C, Traitangwong A, Meeyoo V, Li C S, Zhang S J. Highly active Ni-based catalyst derived from double hydroxides precursor for low temperature CO2 methanation[J]. Ind. Eng. Chem. Res., 2018, 57(28): 9102-9111. doi: 10.1021/acs.iecr.8b01619
Dias Y R, Bernardi F, Perez-Lopez O W. Improving low-temperature CO2 methanation by promoting Ni - Al LDH - derived catalysts with alkali metals[J]. ChemCatChem, 2023, 15(22): e202300834. doi: 10.1002/cctc.202300834
Kathiraser Y, Thitsartarn W, Sutthiumporn K, Kawi S. Inverse NiAl2O4 on LaAlO3 -Al2O3: Unique catalytic structure for stable CO2 reforming of methane[J]. J. Phys. Chem. C, 2013, 117: 8120-8130. doi: 10.1021/jp401855x
Vos B, Poels E, Bliek A. Impact of calcination conditions on the structure of alumina-supported nickel particles[J]. J. Catal., 2001, 198: 77-88. doi: 10.1006/jcat.2000.3082
Li W Y, Zhao G F, Zhong J W, Xie J. Upgrading renewable biogas into syngas via bi-reforming over high-entropy spinel-type catalysts derived from layered double hydroxides[J]. Fuel, 2024, 358: 130155. doi: 10.1016/j.fuel.2023.130155
Casarin M, Falcomer D, Glisenti A, Vittadini A. Experimental and theoretical study of the interaction of CO2 with α-Al2O3[J]. Inorg. Chem., 2003, 42: 436-445. doi: 10.1021/ic0257773
Burger T, Koschany F, Thomys O, Köhler K, Hinrichsen O. CO2 methanation over Fe- and Mn-promoted co-precipitated Ni-Al catalysts: Synthesis, characterization and catalysis study[J]. Appl. Catal. A-Gen., 2018, 558: 44-54. doi: 10.1016/j.apcata.2018.03.021
Weilach C, Spiel C, Föttinger K, Rupprechter G. Carbonate formation on Al2O3 thin film model catalyst supports[J]. Surf. Sci., 2011, 605: 1503-1509. doi: 10.1016/j.susc.2011.05.025
Fottinger K, Schlögl R, Rupprechter G. The mechanism of carbonate formation on Pd-Al2O3 catalysts[J]. Chem. Commun., 2008, 3: 320-322.
Ma Y, Liu J, Chu M, Yue J R, Cui Y B, Xu G W. Enhanced low - temperature activity of CO2 methanation over Ni/CeO2 catalyst[J]. Catal. Lett., 2021, 152: 872-882.
李志红, 黄伟, 左志军, 宋雅君, 谢克昌. 用XPS研究不同方法制备的CuZnAl一步法二甲醚合成催化剂[J]. 催化学报, 2009,30,(2): 171-177. doi: 10.3321/j.issn:0253-9837.2009.02.017LI Z H, HUANG W, ZUO Z J, SONG Y J, XIE K C. XPS study on CuZnAl catalysts prepared by different methods for direct synthesis of dimethyl ether[J]. Chin. J. Catal., 2009, 30(2): 171-177. doi: 10.3321/j.issn:0253-9837.2009.02.017

Figure 5 XRD patterns of calcined (A) and reduced (B) NiAlNd catalysts
For the reduced catalysts, the catalysts were reduced at 500 ℃ for 2 h.

Figure 7 CO2-TPD profiles (A) and the amount of CO2 desorption (B) over the NiAlNd catalysts
Table 1. Texture characteristic parameters of catalysts
| Samples | SBET / (m2·g-1) | Vpore / (cm3·g-1) | Dpore / nm | dNiOa / nm | SNib / (m2·g-1) |
| NiAl | 215 | 0.29 | 5.3 | 3.4 | 23.2 |
| NiAlNd-0.1 | 212 | 0.36 | 6.7 | 3.0 | 24.8 |
| NiAlNd-0.2 | 223 | 0.57 | 10.3 | 2.9 | 27.7 |
| NiAlNd-0.3 | 216 | 0.41 | 7.6 | 2.5 | 21.9 |
| NiAlNd-0.4 | 202 | 0.40 | 8.0 | 2.3 | 21.8 |
| NiAlNd-0.5 | 127 | 0.21 | 6.5 | 1.9 | 10.9 |
| a Determined with Scherrer equation using the diffraction peak at 2θ of 43.4°; b The surface of metallic Ni estimated from CO chemisorption assuming that the ratio of CO to surface Ni atom is 1∶1. | |||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Distribution of metal element in the bulk and on the surface of NiAlNd catalyst
| Sample | Atomic fraction of bulk metala / % | Atomic fraction of surface metalb / % | |||||
| Ni | Al | Nd | Ni | Al | Nd | ||
| NiAl | 70.9(68.0) | 29.1(32.0) | — | 19.9 | 80.1 | 0.0 | |
| NiAlNd-0.1 | 69.2(68.0) | 28.3(28.8) | 2.5(3.2) | 24.3 | 71.6 | 4.1 | |
| NiAlNd-0.2 | 69.1(68.0) | 25.5(25.6) | 5.4(6.4) | 23.4 | 72.2 | 4.4 | |
| NiAlNd-0.3 | 67.2(68.0) | 23.8(22.4) | 9.0(9.6) | 23.2 | 70.9 | 5.9 | |
| NiAlNd-0.4 | 68.2(68.0) | 20.4(19.2) | 11.4(12.8) | 23.7 | 69.0 | 7.2 | |
| NiAlNd-0.5 | 65.7(68.0) | 18.6(16.0) | 15.7(16.0) | 21.6 | 69.3 | 9.1 | |
| a The bulk metal atomic fraction was determined by ICP-AES, and the value in the parenthesis was the nominal value; b The surface metal atomic fraction was determined by XPS. | |||||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们









 下载:
下载: