引用本文:
赵杰, 刘森, 殷齐康, 鲁效庆, 王兆杰. 碱金属修饰的萘炔/萘二炔对CO2选择性吸附分离的理论计算[J]. 无机化学学报,
2024, 40(3): 515-522.
doi:
10.11862/CJIC.20230385 Citation:
Jie ZHAO, Sen LIU, Qikang YIN, Xiaoqing LU, Zhaojie WANG. Theoretical calculation of selective adsorption and separation of CO2 by alkali metal modified naphthalene/naphthalenediyne[J]. Chinese Journal of Inorganic Chemistry,
2024, 40(3): 515-522.
doi:
10.11862/CJIC.20230385
Received Date:
16 October 2023 Revised Date:
12 January 2024 Available Online:
10 March 2024
Abstract:
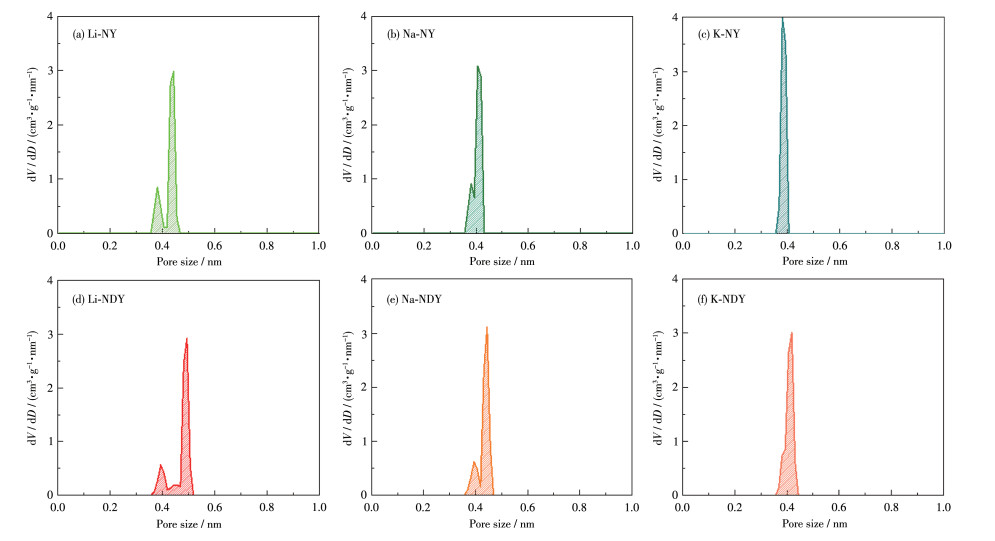
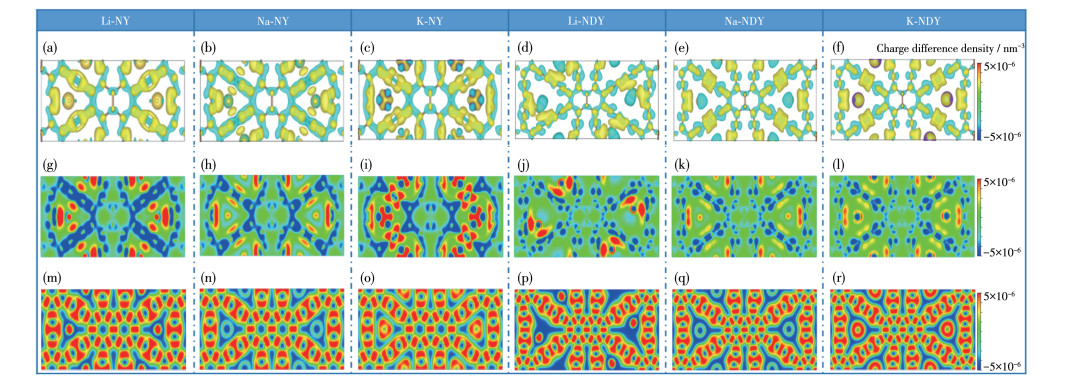
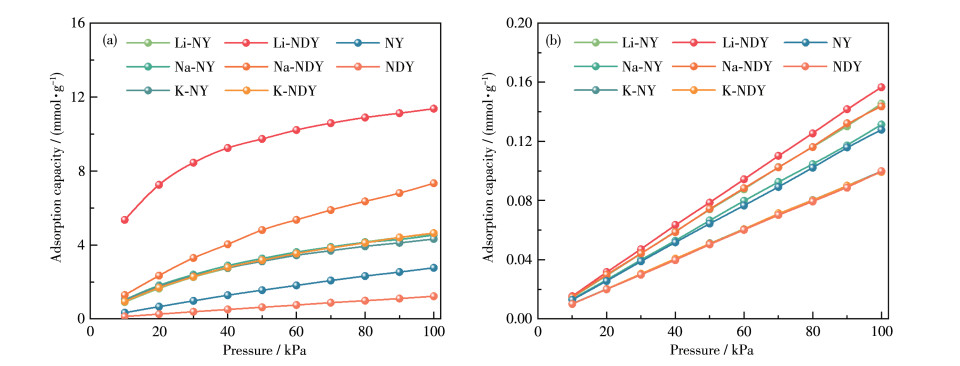
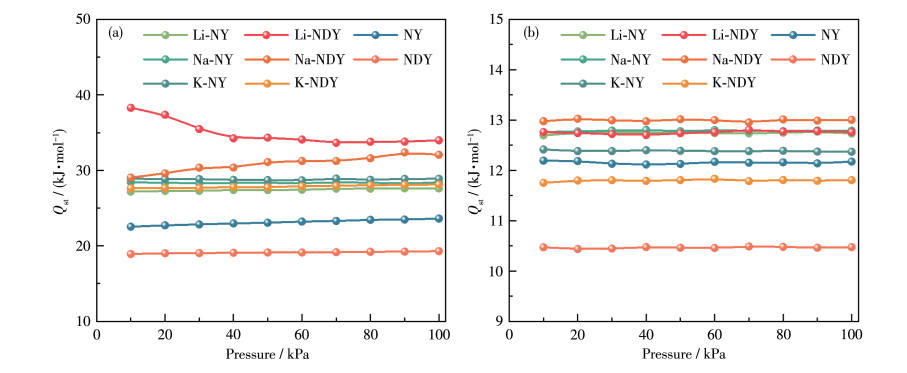
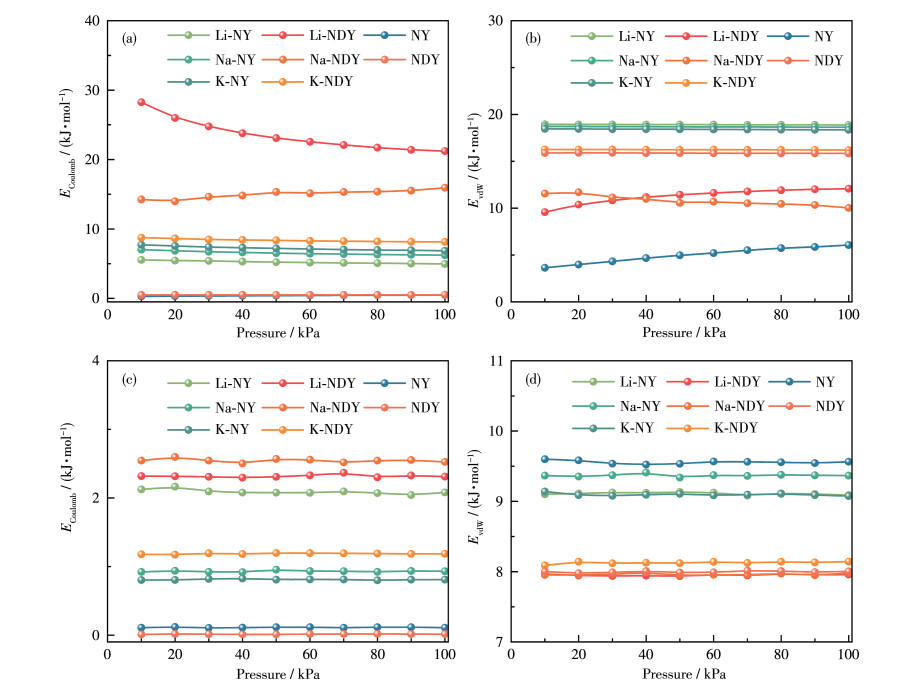
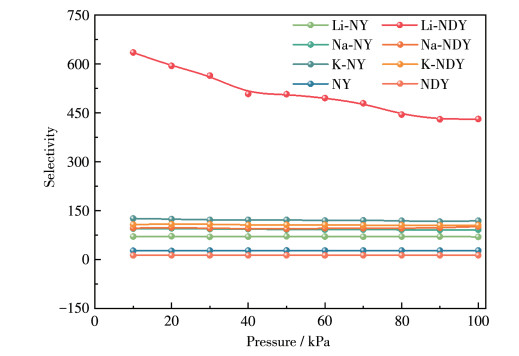
In this study, a combination of grand canonical Monte Carlo and density functional theory was employed to investigate the CO2 adsorption and separation behavior of naphyne (NY) and naphdiyne (NDY) with different alkali metal (AM, including Li, Na, and K) dopants. By analyzing the binding energy, cohesive energy, and electronic properties, it is found that AM-modified NY and NDY exhibit good structural stability. Under conditions of 298 K and 100 kPa, Li-NDY (the NDY modified by Li), exhibits a CO2 adsorption capacity of 11.37 mmol·g-1, with a selectivity for CO2 over N2 of 430.85. Furthermore, the gas adsorption density distribution elucidates the reasons behind the high adsorption capacity of AM-NY and AM-NDY and the inherent difference in their performance. Finally, the modified mechanisms introduced by the AM dopants were discussed in detail from the perspectives of adsorption heat, Coulomb and van der Waals interactions, and other factors.
利用DFT对AM-NY、AM-NDY结构进行优化,并计算原子的电荷[7-8],结构优化时的K点文件分别设定为7×11×1和5×9×1,电荷计算时的K点文件则设定为15×15×1[9]。所有DFT计算均使用Vienna ab initio模拟软件包(VASP)进行[10]。交换关联泛函分别采用PBE(periodic boundary embedding)与GGA(generalized gradient approximation)进行描述[11]。另外通过密度泛函理论色散校正(DFT-D)方法来描述vdW相互作用。采用RASPA对CO2/N2的吸附与分离行为进行了研究[12-13]。CO2和N2分子采用Potoff和Siepmann报道的TraPPE模型进行描述[14]。AM原子和C原子分别采用UFF(universal force field)和Dreiding力场进行描述。在GCMC过程中,采用LJ(Lennard-Jones)势和库仑相互作用分别描述vdW和Ewald静电相互作用,用LJ-12-6势(ULJ)和库仑定律对结构的势能进行了计算,具体公式如下:
Wang M H, Kong L Y, LU X Q, Chi-Man L W. First-row transition metal embedded pyrazine-based graphynes as high-performance single atom catalysts for the CO2 reduction reaction[J]. J. Mater. Chem. A,
2022, 10(16):
9048-9058.
doi: 10.1039/D2TA00654E
[2]
Song K S, Fritz P W, Coskun A. Porous organic polymers for CO2 capture, separation and conversion[J]. Chem. Soc. Rev.,
2022, 51(23):
9831-9852.
doi: 10.1039/D2CS00727D
[3]
Yu Y S, Zhang C, Ding W C, Zhang Z X, Wang G X. Determining the performance for an integrated process of COD removal and CO2 capture[J]. J. Clean. Prod.,
2020, 275:
122845.
doi: 10.1016/j.jclepro.2020.122845
[4]
Zhou S N, Wang M H, Wei S X, Cao S F, Wang Z J, Liu S Y, Sun D F, Lu X Q. First-row transition-metal-doped graphyne for ultrahigh-performance CO2 capture and separation over N2/CH4/H2[J]. Mat. Today Phys.,
2021, 16:
100301.
doi: 10.1016/j.mtphys.2020.100301
[5]
Liu Y, Liu W B, Wang R G, Hao L F, Jiao W C. Hydrogen storage using Na-decorated graphyne and its boron nitride analog[J]. Int. J. Hydrogen Energ.,
2014, 39(24):
12757-12764.
doi: 10.1016/j.ijhydene.2014.06.107
[6]
Zhou S N, Wang M H, Wei S X, Xin H L, Zhai W R, Xu S Y, Liu S, Liu S Y, Wang Z J, Chi-Man L W, Lu X Q. Multi-objective optimization of alkali/alkaline earth metals doped graphyne for ultrahigh-performance CO2 capture and separation over N2/CH4[J]. Mat. Today Phys.,
2021, 21:
100539.
doi: 10.1016/j.mtphys.2021.100539
[7]
Marsusi F, Drummond N D, Verstraete M J. The physics of single-side fluorination of graphene: DFT and DFT+U studies[J]. Carbon,
2019, 144:
615-627.
doi: 10.1016/j.carbon.2018.12.089
[8]
Jagiello J, Thommes M. Comparison of DFT characterization methods based on N2, Ar, CO2, and H2 adsorption applied to carbons with various pore size distributions[J]. Carbon,
2004, 42(7):
1227-1232.
doi: 10.1016/j.carbon.2004.01.022
[9]
Guo C, Zhang T, Deng X X, Liang X Y, Guo W Y, Lu X Q, Chi-Man L W. Electrochemical CO2 reduction to C1 products on single nickel/cobalt/iron‑doped graphitic carbon nitride: A DFT study[J]. ChemSusChem,
2019, 12(23):
5126-5132.
doi: 10.1002/cssc.201902483
[10]
Kresse G, Furthmüller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Phys. Rev. B,
1996, 54(16):
11169-11186.
doi: 10.1103/PhysRevB.54.11169
[11]
Luo K, Karasiev V V, Trickey S B. A simple generalized gradient approximation for the noninteracting kinetic energy density functional[J]. Phys. Rev. B,
2018, 98(4):
041111.
doi: 10.1103/PhysRevB.98.041111
[12]
Dubbeldam D, Calero S, Ellis D E, Snurr R Q. RASPA: Molecular simulation software for adsorption and diffusion in flexible nanoporous materials[J]. Mol. Simulat.,
2016, 42(2):
81-101.
doi: 10.1080/08927022.2015.1010082
[13]
Gupta A, Chempath S, Sanborn M J, Clark L A, Snurr R Q. Object-oriented programming paradigms for molecular modeling[J]. Mol. Simul.,
2003, 29(1):
29-46.
doi: 10.1080/0892702031000065719
[14]
Potoff J J, Siepmann J I. Vapor-liquid equilibria of mixtures containing alkanes, carbon dioxide, and nitrogen[J]. AIChE J.,
2001, 47(7):
1676-1682.
doi: 10.1002/aic.690470719
[15]
Xu S Y, Wei S X, Wang L, Liu S, Wang M H, Liu S Y, Wang Z J, Yang T F, Lu X Q. Li-decorated β1-graphyne for high-performance CO2 capture and separation over N2[J]. Appl. Surf. Sci.,
2022, 605:
154724.
doi: 10.1016/j.apsusc.2022.154724
[16]
Liu C, Liu Z X, Ye X J, Cheng P, Li Y J. First-principles study of structural, elastic and electronic properties of naphyne and naphdiyne[J]. RSC Adv.,
2020, 10(58):
35349-35355.
doi: 10.1039/D0RA07214A
[17]
Li Y J, Li Y Y, Lin P, Gu J, He X J, Yu M X, Wang X T, Liu C, Li C X. Architecture and electrochemical performance of alkynyl-linked naphthyl carbon skeleton: Naphyne[J]. ACS Appl. Mater. Interfaces,
2020, 12(29):
33076-33082.
doi: 10.1021/acsami.0c05741
[18]
Tao L, Huang J C, Dastan D, Wang T Y, Li J, Yin X T, Qi W. CO2 capture and separation on charge-modulated calcite[J]. Appl. Surf. Sci.,
2020, 530:
147265.
doi: 10.1016/j.apsusc.2020.147265
[19]
Yan B L, Yu S, Zeng C F, Yu L, Wang C Q, Zhang L X. Binderless zeolite NaX microspheres with enhanced CO2 adsorption selectivity[J]. Microporous Mesoporous Mat.,
2019, 278:
267-274.
doi: 10.1016/j.micromeso.2018.12.002
[20]
Balbuena P B, Gubbins K E. Theoretical interpretation of adsorption behavior of simple fluids in slit pores[J]. Langmuir,
1993, 9(7):
1801-1814.
doi: 10.1021/la00031a031
[21]
Wang L, Zhao J J, Wang L L, Yan T Y, Sun Y Y, Zhang S B. Titanium-decorated graphene oxide for carbon monoxide capture and separation[J]. Phys. Chem. Chem. Phys.,
2011, 13(47):
21126-21131.
doi: 10.1039/c1cp21778j
[22]
Suraweera N S, Albert A A, Peretich M E, Abbott J, Humble J R, Barnes C E, Keffer D J. Methane and carbon dioxide adsorption and diffusion in amorphous, metal-decorated nanoporous silica[J]. Mol. Simulat.,
2014, 40(7/8/9):
618-633.
[23]
Tian Z H, Huang J J, Zhang X, Shao G L, He Q Y, Cao S K, Yuan S G. Ultra-microporous N-doped carbon from polycondensed framework precursor for CO2 adsorption[J]. Microporous Mesoporous Mat.,
2018, 257:
19-26.
doi: 10.1016/j.micromeso.2017.08.012
[24]
Lekien F, Marsden J. Tricubic interpolation in three dimensions[J]. Int. J. Numer. Methods Eng.,
2005, 63(3):
455-471.
doi: 10.1002/nme.1296
[25]
Torres-Knoop A, Balaji S P, Vlugt T J H, Dubbeldam D. A comparison of advanced monte Carlo methods for open systems: CFCMC vs CBMC[J]. J. Chem. Theory Comput.,
2014, 10(3):
942-952.
doi: 10.1021/ct4009766
[26]
Liu A Q, Peng X, Jin Q B, Jain S K, Vicent-Luna J M, Calero S, Zhao D F. Adsorption and diffusion of benzene in Mg-MOF-74 with open metal sites[J]. ACS Appl. Mater. Interfaces,
2019, 11(4):
4686-4700.
doi: 10.1021/acsami.8b20447
[27]
Yu J M, Xie L H, Li J R, Ma Y G, Seminario J M, Balbuena P B. CO2 capture and separations using MOFs: Computational and experimental studies[J]. Chem. Rev.,
2017, 117(14):
9674-9754.
doi: 10.1021/acs.chemrev.6b00626
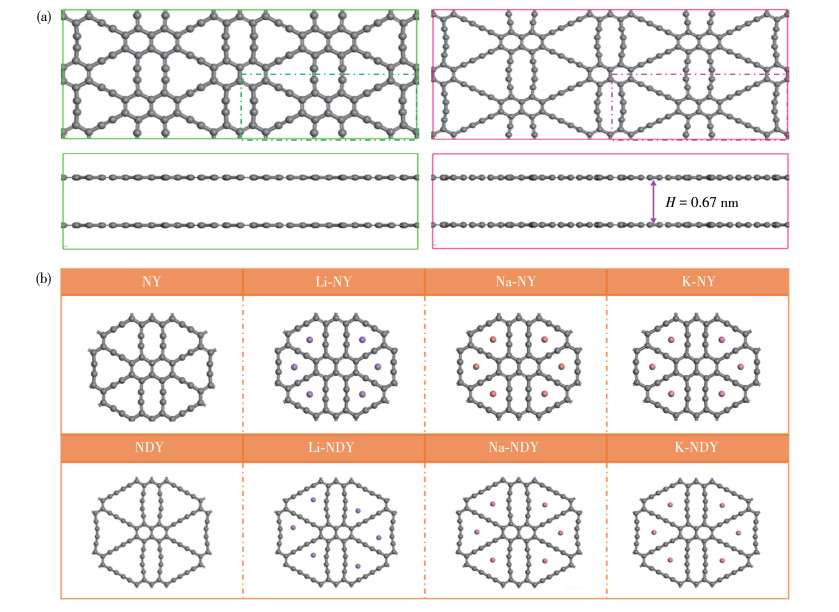
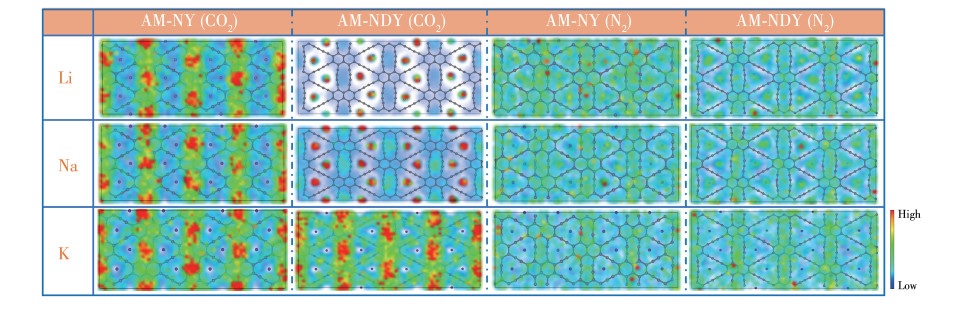
图 1
二维NY和NDY (a)及AM修饰二维NY和NDY (b)的模型
Figure 1
Models of two-dimensional NY and NDY (a), and AM-modified two-dimensional NY and NDY (b)

 下载:
下载:

 下载:
下载:









 下载:
下载:
