Table 1.
Crystal data and structure refinements of the compounds
Citation:
Mengru CAO, Guoyuan JIANG, Hongli LI, Sinong LI, Huihua SONG. Two pairs of chiral cobalt enantiomeric coordination compounds based on D-(-)-/L-(+)-4-hydroxyphenylglycine: Synthesis, crystal structures, and electrochemical recognition[J]. Chinese Journal of Inorganic Chemistry,
2024, 40(1): 232-246.
doi:
10.11862/CJIC.20230381

基于D-(-)-/L-(+)-对羟基苯甘氨酸的两对手性钴配合物的合成、晶体结构和电化学识别
摘要:
采用常温溶液挥发法,以D-(—)-/L-(+)-对羟基苯甘氨酸(D-/L-Hhpg)为主配体,2种含氮吡啶配体4,4′-联吡啶(4,4′-bipy)和5,5′-二甲基-2,2′-联吡啶(5,5′-BM-2,2′-bipy)为辅助配体,与CoCl2·6H2O反应合成了2对手性配合物{[Co (D-hpg)(4,4′-bipy)(H2O)]Cl·H2O}n(1-D)、{[Co (L-hpg)(4,4′-bipy)(H2O)]Cl·H2O}n(1-L)、[Co (D-hpg)2(5,5′-BM-2,2′-bipy)]Cl·5.5H2O (2-D)、[Co (L-hpg)2(5,5′-BM-2,2′-bipy)]Cl·5.5H2O (2-L)。通过单晶X射线衍射、元素分析、红外光谱、粉末X射线衍射等多种测试方法对其结构进行分析和表征。配合物的单晶X射线衍射数据表明,配合物1-D和1-L属于单斜晶系,P21手性空间群,分别呈现1D左手螺旋链和右手螺旋链,通过4,4′-bipy分子扩展为2D网状矩形格子结构。配合物2-D属于单斜晶系,P21手性空间群,为0D小分子,在氢键的作用下形成1D超分子双链,并以ABAB形式在a轴方向堆积排布。这些配合物的结构差异归因于辅助配体和Hhpg配位模式的影响。此外,还研究了配合物的电化学性质。配合物1-D表现出电化学可逆的氧化还原行为,并可作为电化学传感器用于有效地检测组氨酸对映体和定量测定组氨酸混合物中的对映体过量。
-
关键词:
- D-(-)-/L-(+)-对羟基苯甘氨酸
- / 手性配合物
- / 晶体结构
- / 电化学识别
English
Two pairs of chiral cobalt enantiomeric coordination compounds based on D-(-)-/L-(+)-4-hydroxyphenylglycine: Synthesis, crystal structures, and electrochemical recognition
Abstract:
Two pairs of chiral coordination compounds {[Co(D-hpg)(4,4′-bipy)(H2O)]Cl·H2O}n (1-D), {[Co(L-hpg)(4,4′-bipy)(H2O)]Cl·H2O}n (1-L), [Co(D-hpg)2(5,5′-BM-2,2′-bipy)]Cl·5.5H2O (2-D), and [Co(L-hpg)2(5,5′-BM-2,2′-bipy)] Cl·5.5H2O (2-L), where D-Hhpg=D-(-)-4-hydroxyphenylglycine, L-Hhpg=L-(+)-4-hydroxyphenylglycine, 4,4′-bipy= 4,4′-bipyridine, 5,5′-BM-2,2′-bipy=5,5′-dimethyl-2,2′-bipyridine, have been successfully synthesized. Their structures were determined by single-crystal X-ray diffraction analysis and characterized by elemental analysis, X-ray photoelectron spectra, infrared spectroscopy, solid-state circular dichroism spectra, thermogravimetric analysis, powder X-ray diffraction, and electrochemical methods. Compounds 1-D and 1-L feature 2D (4, 4) rectangular grid networks that consist of left-or right-handed helical chains. Compounds 2-D and 2-L exhibit 0D molecule structures, and further 1D supramolecular double chains are formed by hydrogen bonding. The differences in the structures of these compounds are attributed to the influence of ancillary N-donor ligands and the coordination modes of Hhpg. Moreover, compound 1-D displays electrochemically reversible redox behavior, and serves as an electrochemical sensor for efficiently detecting histidine (His) enantiomers and quantitatively determining the enantiomeric excess in the His mixture.
-
0. Introduction
Chirality is a universal aspect of the chemical sciences[1-2]. Recognition of chiral amino acids plays a pivotal part in chemical biology and pharmacology, since different enantiomeric amino acids may make quite a difference in biological behavior and pharmacological activity[3-6]. L-histidine (L-His), as a semi-essential amino acid, can alleviate some physiological illnesses such as heart disease, anemia, and rheumatoid arthritis, while D-histidine (D-His) has no nutritional value to the human body. It is often used in scientific research to study the structure-function analysis of enzymes[7-11]. Thus, exploiting effective and practical techniques for chiral analysis of His enantiomers in the biochemical analysis is meaningful. Conventional detection technologies for His enantiomers mainly include high-performance liquid chromatography, fluorescence, etc[12-13]. However, these methods require high concentrations of analytes, sophisticated operation, and relatively expensive instrumentation, and HPLC is also typically time-consuming[14]. In this context, the highly selective, low-cost, fast-speed, real-time, and online operation electrochemical sensors have been evaluated as a promising chiral analysis technique[15-18].
The key to electrochemical chiral sensing is the material used to prepare the sensor′s electroactive surface. The chiral coordination compounds (CCPs) are an intriguing class of crystalline materials formed usually by the self-assembly of metal ions and chiral polydentate ligands[19-21]. These materials are highly promising for electrochemical chiral sensors owing to the ultrahigh surface area, precise network structures, and their exposed active sites of ligand groups and metal ions that provide selective recognition and boost the charge transports[22]. So far, electrochemical methods based on CCPs have had limited reports for the identification of chiral isomers because few applications were developed based on the current difference in voltammetry. For instance, Rufina A. Zilberg et al. synthesized mixed chelate compounds [M(S-Ala)2(H2O)n][M(S-Phe)2(H2O)n] (M=Cu(Ⅱ), Zn(Ⅱ); n=0-1), which can be used to analyze Nap and Prp enantiomers[23]. Therefore, CCPs are a promising electrode modification material for electrochemical chiral recognition.
Among the synthetic methods of chiral coordination compounds, using the chiral ligands is the most direct and effective method[24-31]. Amino acids and their derivatives have both amino and carboxyl functional groups, which exhibit excellent coordination capabilities. The carbonyl oxygen atoms, hydroxyl oxygen atoms, and nitrogen atoms can form hydrogen bonds and further form supramolecular structures. Chiral amino acid derivatives have also been confirmed to have some contributions in the research of recognition performance[32]. D-(-)-/L-(+)-4-Hydroxyphenylglycine (D-/L-Hhpg) as amino acid derivative chiral ligands have natural advantages due to their being cheap, easily available, non-toxic, and harmless. We adopt a mixed-ligand strategy that selects D-Hhpg and L-Hhpg as chiral ligands with N-donor ligands and transition Co(Ⅱ) metal ions to construct chiral coordination compounds.
With this background information, herein two pairs of chiral coordination compounds {[Co(D-hpg)(4,4′-bipy)(H2O)]Cl·H2O}n (1-D), {[Co(L-hpg)(4,4′-bipy)(H2O)]Cl·H2O}n (1-L), [Co(D-hpg)2(5,5′-BM-2,2′-bipy)]Cl·5.5H2O (2-D), and [Co(L-hpg)2(5,5′-BM-2,2′-bipy)]Cl·5.5H2O (2-L), where 4,4′-bipy=4,4′-bipyridine, 5,5′-BM-2,2′-bipy=5,5′-dimethyl-2,2′-bipyridine, have been successfully synthesized and characterized by elemental analysis, X-ray photoelectron spectroscopy (XPS), infrared spectroscopy, thermogravimetric analysis (TGA), powder X-ray diffraction (PXRD), and single-crystal X-ray diffraction. The effects of N-donor ligands on the structures of the compounds have been discussed in detail. Their electrochemical behaviours and electrochemical recognition were further studied via cyclic voltammetry (CV) and differential pulse voltammetry (DPV). Notably, compound 1-D behaved high electroconducting and distinctive oxidation signals for each enantiomer of D-/L-His. Not only is the sensor able to discriminate enantioselectively, but also it allows for the rapid quantitative determination of enantiomeric excess in the His mixture. Compared with other materials requiring multistep syntheses, it is an extraordinary and meaningful work to use cheap and easy synthesized crystals for enantiomeric identification.
1. Experimental
1.1 Material and measurements
All chemicals and solvents in the syntheses were purchased from commercial sources and were used as received without further purification. IR spectra were collected on an FTIR-8900 spectrometer within the 4 000-400 cm-1 region (KBr pellets). TGA experiments were carried out on a simultaneous STA 449F3/TENSOR 27 thermal analyzer under a static N2 atmosphere with a heating rate of 10 ℃·min-1 from room temperature to 800 ℃. Elemental analyses were performed on an Elemental Vario EL elemental analyzer. XPS spectrum was recorded on a Thermascientific ESCALAB QXi instrument with Al Kα X-rays as the excitation source. The PXRD patterns were obtained on a Bruker D8-Advance X-ray diffractometer with Cu Kα radiation (λ=0.154 2 nm, U=40 kV, I=40 mA) in a 2θ range of 5°-50° at room temperature. The solid-state circular dichroism (CD) spectra were recorded on a JASCO J-810 spectropolarimeter with KCl pellets. UV-Vis spectra were acquired with a Shimadzu 2501 spectrometer (Japan). CV and DPV measurements were carried out on a CHI660 electrochemical workstation (Shanghai Chen Hua Instruments, China) at room temperature. A traditional three-electrode system consisting of a glassy carbon electrode (GCE) as the working electrode, a saturated calomel electrode (Ag/AgCl) as the reference electrode, and a platinum wire as the counter electrode was used.
1.2 Synthesis
1.2.1 Synthesis of compound 1-D
CoCl2·6H2O (0.047 6 g, 0.2 mmol), D-Hhpg (0.016 7 g, 0.1 mmol), and distilled water (10 mL) were mixed and kept stirring until completely dissolved, and then the methanol solution (6 mL) with 4,4′-bipy (0.015 6 g, 0.1 mmol) was added to the above solution and kept stirring for 20 min. The resulting solution was filtered and the filtrate was slowly evaporated at room temperature for 5-7 d or so to deposit red flaky crystals of compound 1-D with 55% yield based on Co. Anal. Calcd. for C18H20N3ClCoO5(%): C, 47.75; H, 4.45; N, 9.28. Found(%): C, 47.18; H, 4.44; N, 9.38. IR (KBr, cm-1): 3 364 (s), 3 318(w), 1 608 (s), 1 589 (s), 1 498 (m), 1 413 (s), 1 212 (m), 1 133 (s), 1 055 (m), 1 010 (m), 808 (s), 763 (w), 731 (m), 620 (s), 575 (w).
1.2.2 Synthesis of compound 1-L
The synthesis method of 1-L was the same as 1-D, and the difference was to replace D-Hhpg with L-Hhpg. Compound 1-L is red flaky crystals that were produced in a yield of 61% based on Co. Anal. Calcd. for C18H20N3ClCoO5(%): C, 47.75; H, 4.45; N, 9.28. Found: C, 47.09; H, 4.39; N, 9.80. IR (KBr, cm-1): 3 363 (s), 3 305(w), 1 615 (s), 1 562 (s), 1 504 (m), 1 411 (s), 1 219 (m), 1 099 (s), 1 045 (m), 1 017 (m), 818 (s), 765 (w), 735 (m), 620 (s), 575 (w). The XPS spectra of Co2p are shown in Fig.S1 (Supporting information). For compound 1-L, there were two main peaks corresponding to Co2p3/2 and Co2p1/2 at 780.82 and 796.36 eV, respectively. The peaks at 785.60 eV and 802.34 eV are the satellite peaks of Co2p3/2 and Co2p1/2, respectively. According to bond valence calculation (BVS) and the law of conservation of charge, the central metal ion in compound 1-L is Co(Ⅱ).
1.2.3 Synthesis of compound 2-D
CoCl2·6H2O (0.047 6 g, 0.2 mmol), D-Hhpg (0.016 7 g, 0.1 mmol), and distilled water (8 mL) were mixed and kept stirring until completely dissolved, and then the ethanol solution (8 mL) with 5,5′-BM-2,2′-bipy (0.018 4 g, 0.1 mmol) was added to the above solution and kept stirring for 20 min. The resulting solution was filtered and the filtrate was slowly evaporated at room temperature for two weeks or so to deposit red block crystals of compound 2-D with 65% yield based on Co. Anal. Calcd. for C28H39ClCoN4O11.5(%): C, 47.37; H, 5.54; N, 7.89. Found(%): C, 47.46; H, 5.78; N, 7.86. IR (KBr, cm-1): 3 437 (s), 3 193(m), 3 084(w), 1 662 (s), 1 513 (m), 1 475 (m), 1 347 (m), 1 244 (s), 1 173 (w), 1 046 (w), 834 (m), 724 (w), 625(w), 574 (m). The XPS spectra of Co2p are shown in Fig.S1. For 2-D, there were two main peaks corresponding to Co2p3/2 and Co2p1/2 at 781.01 and 796.49 eV, respectively. The peaks at 784.80 eV and 800.44 eV are the satellite peaks of Co2p3/2 and Co2p1/2, respectively. According to BVS and the law of conservation of charge, the central metal ion in compound 2-D is Co(Ⅲ).
1.2.4 Synthesis of compound 2-L
The synthesis method of 2-L was the same as 2-D, and the difference was to replace D-Hhpg with L-Hhpg. Compound 2-L is red block crystals that were produced in a yield of 61% based on Co. Anal. Calcd. for C28H39ClCoN4O11.5(%): C, 47.37; H, 5, 54; N, 7.89. Found(%): C, 47.25; H, 5.82; N, 7.95. IR (KBr, cm-1): 3 430 (s), 1 661 (s), 1 519 (m), 1 448 (w), 1 450(w), 1 357(m), 1 308 (w), 1 260 (s), 1 170(m), 1019 w), 827 (w), 584 (m).
1.2.5 Preparation of working electrodes modified with compounds 1 and 2
Before each experiment, the GCE (diameter: 3 mm) was polished on a polishing pad with 0.3 and 0.05 μm alumina powders. The cleaned GCE was dried with a nitrogen stream for the next modification. 20.0 mg (or 12 mg) crystals were first mixed and ground with carbon black under mass ratios of 1∶3, and then dispersed into 200 μL of 0.5% Nafion solution and ultrasonicated for 120 min to obtain a uniformly dispersed solution. Then, 10 μL catalyst suspension was dropped on the surface of GCEs and dried for testing. The Nafion solution acts as an adhesive and the protecting agent to ensure that the hybrid catalysts can be tightly modified on the surface of the GCE.
1.3 X-ray crystallography
Suitable single crystals for compounds 1 and 2 were selected for single-crystal X-ray diffraction analyses. The data for 1 and 2 were collected on a Bruker SMART-CCD diffractometer with Mo Kα radiation source (λ=0.071 073 nm) by φ-ω scan mode. The structures were solved through direct methods using SHELXS-97 and all non-hydrogen atoms were refined anisotropically by full-matrix least-squares on F2 using SHELXL-97[33-34]. Solvent removal can be carried out by using the SQUEEZE program. Further crystallographic data and experimental details for structural analyses of compounds 1 and 2 are summarized in Table 1. The selected bond lengths and angles of 1 and 2 are given in Table S1. Hydrogen bond lengths and bond angles are collated in Table S2.
Table 1

 下载:
导出CSV
下载:
导出CSV
Parameter 1-D 1-L 2-D Empirical formula C18H20N3ClCoO5 C18H20N3ClCoO5 C28H39ClCoN4O11.5 T/K 298(2) 298(2) 298(2) Formula weight 452.75 452.75 709.92 Space group P21 P21 P21 Crystal system Monoclinic Monoclinic Monoclinic a/nm 1.022 64(13) 1.011 47(9) 1.083 99(11) b/nm 0.909 25(11) 0.902 04(8) 1.353 46(14) c/nm 1.153 49(15) 1.142 67(11) 2.186 3(2) β/(°) 111.806(4) 111.754(4) 91.775(3) V/nm3 0.995 8(2) 0.968 31(16) 3.206 1(6) Z 2 2 4 Dc/(Mg·m-3) 1.510 1.553 1.266 F(000) 466 466 1 264 θ range/(°) 1.902-25.009 2.168-25.016 2.07-29.13 Reflection collected 6 348 5 108 54 80 Independent reflection 2 728 (Rint=0.071 4) 3 043 (Rint=0.052 5) 4 792 (Rint=0.053 2) Data, restraint, number of parameters 2 728, 265, 206 3 043, 1, 212 14 629, 241, 725 Goodness-of-fit on F2 1.236 1.092 1.032 R1, wR2 [I > 2σ(I)] 0.102 1, 0.227 0 0.094 4, 0.203 3 0.070 0, 0.126 9 R1, wR2 (all data) 0.143 5, 0.248 2 0.139 1, 0.224 1 0.129 5, 0.143 1 Flack parameter -0.06(7) 0.05(7) 0.00(3) 2. Results and discussion
2.1 Structure description
2.1.1 Structural description of compounds 1-D and 1-L
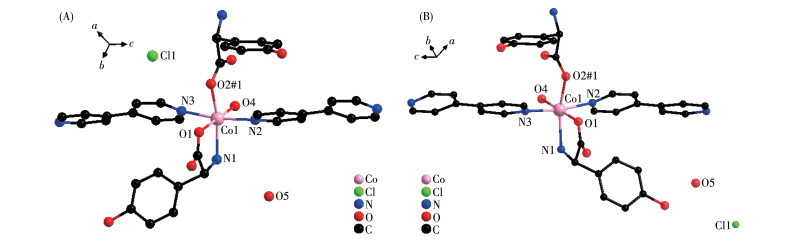
Single crystal X-ray diffraction analysis revealed that compound 1-D crystallizes in the monoclinic space group of P21 and possesses a 2D rectangular grid network. As shown in Fig. 1A, the asymmetric unit is composed of one Co(Ⅱ) cation, one D-hpg- anion, one 4,4′-bipy ligand, one Cl- counter anion, one coordination water molecule, and one lattice water molecule. The Co(Ⅱ) center is coordinated by three oxygen atoms and three nitrogen atoms to form a distorted octahedron geometry (CoO3N3). O1, O2#1, N1 are from two D-hpg- ligands, N2, N3 are from two 4,4′-bipy auxiliary ligands, O4 is from a coordinated water molecule, where the octahedral equatorial positions are O1, O2, O4 and N1, N2 and N3 are in the axial positions of the octahedron. The Co—O bond lengths are 0.205 5(14)-0.213 0(11) nm and the Co—N distances are 0.218 1(15)-0.222 0(11) nm, respectively. All of which fall within the normal ranges[35].
Figure 1
 Figure 1. Coordination environment of Co(Ⅱ) in compounds 1-D (A) and 1-L (B)
Figure 1. Coordination environment of Co(Ⅱ) in compounds 1-D (A) and 1-L (B)H atoms have been omitted for clarity.
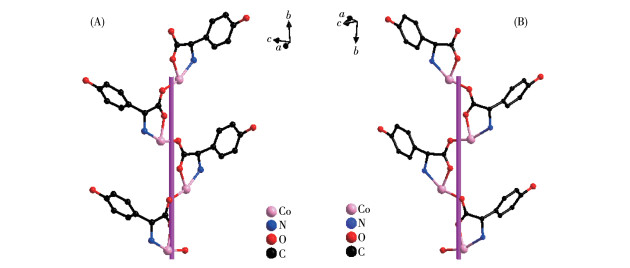
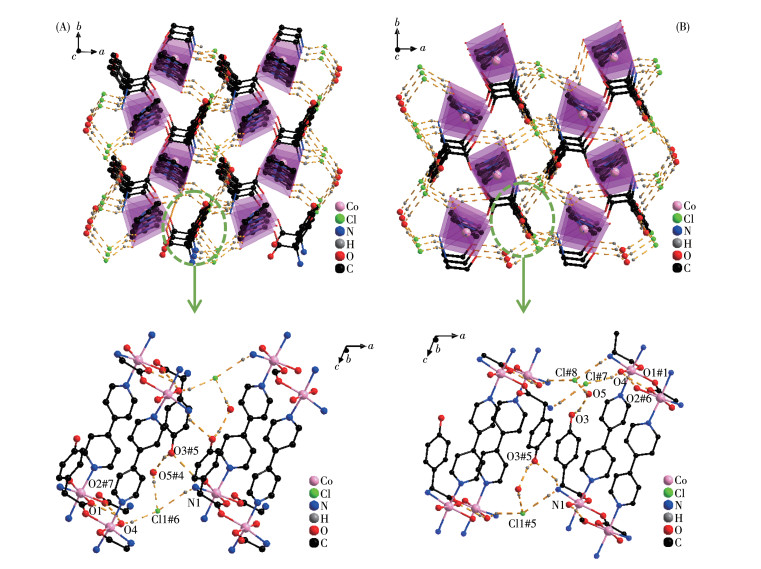
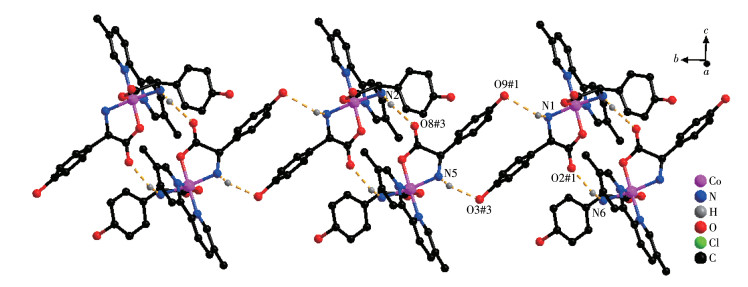
The D-hpg- adopts as a μ2-η1∶η1∶η1 coordination mode (two carboxyl oxygen atoms and one amino nitrogen atom) to connect the two metal cobalt, forming a 1D right-handed helical chain structure along the a-axis (Fig. 2A). In addition, these chains are further linked via 4,4′-bipy molecules to build a 2D (4, 4) rectangular grid network parallel to the bc plane, and the size of the rectangular grid is 0.519 nm×1.153 nm, respectively, as shown in Fig. 3A. There are extensive hydrogen bonds among carboxyl oxygen atoms (O1, O2#7), amino nitrogen atom (N1), coordinated water molecule (O4) and free Cl- anion, which further connects the 2D networks to form a 3D supramolecular structure, as depicted in Fig. 4A. The typical hydrogen bonds are N1⋯O3#5 0.319 0(18) nm, N1⋯Cl1#6 0.340 0(13) nm, O3⋯O5#4 0.272(2) nm, O4⋯O1#1 0.275 3(16) nm, O4⋯O2#7 0.335 2(16) nm, O5⋯Cl1#6 0.320 3(18) nm (Symmetry codes: #1: -x+1, y-1/2, -z+1; #3: -x+1, y+1/2, -z+1; #4: x, y, z-1; #5: -x, y-1/2, -z; #6: x-1, y, z; #7: x, y-1, z).
Figure 2
 Figure 2. (A) One-dimensional right-handed helical chains of compound 1-D; (B) 1D left-handed helical chains of compound 1-L
Figure 2. (A) One-dimensional right-handed helical chains of compound 1-D; (B) 1D left-handed helical chains of compound 1-LFigure 3
Figure 4
By comparing the crystallography data of 1-D and 1-L shown in Table 1 as well as Fig. 1A and 1B, we conclude that compounds 1-D and 1-L are enantiomers. In compound 1-L, the L-hpg- anion adopts the μ2-η1∶η1∶η1 coordination mode to connect adjacent Co ions. Compared with compound 1-D, 1-L forms 1D left-handed helical chains (Fig. 2B). These chains are further linked via 4,4′-bipy molecules to build a 2D (4, 4) rectangular grid network parallel to the bc plane (Fig. 3B), and the size of the rectangular grid is 0.519 nm×1.142 nm, respectively, and 1-L eventually forms a 3D supramolecular structure through hydrogen bond interactions, as depicted in Fig. 4B. The typical hydrogen bonds are N1⋯Cl1#5 0.335 0(13) nm, N1⋯O3#5 0.313 1(17) nm, O3⋯O5 0.267(2) nm, O4⋯O1#1 0.270 8(16) nm, O4⋯O2#6 0.332 2(15) nm, O4⋯Cl1#7 0.309 5(12) nm (Symmetry codes: #1: -x+1, y+1/2, -z+2; #5: -x, y+1/2, -z+1; #6: x, y+1, z; #7: x, y+1, z+1).
2.1.2 Crystal structure of compound 2-D
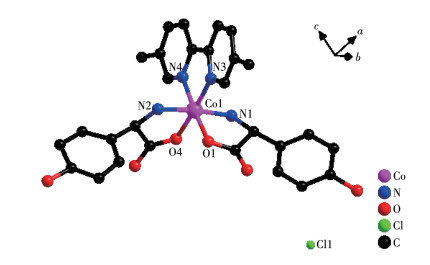
Single crystal X-ray diffraction analysis revealed that compound 2-D crystallizes in a monoclinic space group of P21 and possesses a 0D small molecule structure. As shown in Fig. 5, the asymmetric unit is composed of one Co(Ⅲ) cation, two D-hpg- anions, one 5,5′-BM-2,2′-bipy ligand, one Cl- counter anion, and 5.5 lattice water molecules. Each Co(Ⅲ) is six-coordinated with the geometry of distorted octahedral geometry, which is completed by O1, N1, N2, O4 from two D-hpg- ligands and N3, N4 from one 5,5′-BM-2,2′-bipy ligand. The O1, O4, N2, and N3 atoms form the equatorial plane, while the N1 and N2 atoms are located in the axial positions. The Co—O bond lengths are 0.184 2(7) and 0.187 8(6) nm, and the Co—N bond lengths range from 0.188 2(8) to 0.193 7(9) nm, which are in accordance with the previously reported compounds[35].
Figure 5
 Figure 5. View of coordination environment of Co(Ⅱ) in compound 2-D
Figure 5. View of coordination environment of Co(Ⅱ) in compound 2-DH atoms are omitted for clarity.
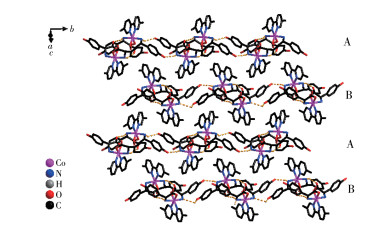
Both the two D-hpg- anions and 5,5′-BM-2,2′-bipy ligands act as bidentate ligands with chelating mode (one carboxyl oxygen atom and one amino nitrogen atom, two pyridine nitrogen atoms) to coordinate with the metal cobalt ions, so each asymmetric unit contains three penta-atomic chelate rings. However, adjacent small molecules are connected by hydrogen bonds between the carboxyl oxygen atoms and amino nitrogen atoms of the D-hpg- ligand to form a dimer unit. These dimer units form a 1D supramolecular double chain along the b-axis through the hydrogen bonds between the hydroxyl oxygen atom and the amino nitrogen atom of the D-hpg- ligand (the hydrogen bonds: N2⋯O8#3 0.297 8(11) nm, N6⋯O(2)#1 0.284 2(11) nm, N1⋯O9#1 0.311 0(12) nm, N5⋯O3#3 0.295 4(13) nm), as shown in Fig. 6. In addition, the 1D double chains are arranged in the form of ABAB stacking in the a-axis direction, as shown in Fig. 7. The typical hydrogen bonds are N1⋯O9#1 0.311 0(12) nm, N1⋯Cl2#2 0.330 2(8) nm, N2⋯O8#3 0.297 8(11) nm, N5⋯Cl1# 0.319 1(9) nm, N5⋯O3#3 0.295 4(13) nm, N6⋯O2#1 0.284 2(11) nm, O9⋯N1#3 0.311 0(12) nm (Symmetry codes: #1: -x, y+1/2, -z+1; #2: x-1, y, z; #3: -x, y-1/2, -z+1).
Figure 6
Figure 7
2.1.3 Comparison of the structures
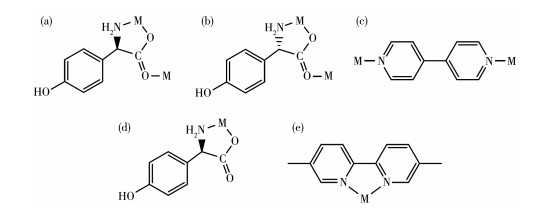
From the structure descriptions above, we found the different auxiliary ligands (4,4′-bipy, 5,5′-BM-2,2′-bipy) affected the coordination mode of the organic ligands, further, the structure of compounds 1 and 2 were transformed (Scheme 1). When the 4,4′-bipy molecule is selected as the auxiliary ligand in 1, the D/L-hpg- exhibits the μ2-η1∶η1∶η1 coordination mode and bridges the more neighboring Co(Ⅱ) cations and extends infinitely along the b-axis direction to form a 1D helical chain. These 1D chains are further linked via 4,4′-bipy molecules to build a 2D rectangular grid network. When 5,5′-BM-2,2′-bipy is used instead of 4,4′-bipy in 2-D, the D-Hhpg and L-Hhpg ligands exhibit chelating bidentate coordination patterns with Co(Ⅲ) ions. Moreover, the 5,5′-BM-2,2′-bipy molecule also participates in the coordination of compound 2-D through two pyridine nitrogen atoms in a chelated bidentate mode, which does not act as a bridge, and the compound finally presents a 0D molecular structure. In short, different N-donor ligands induce a shift in the coordination mode of Hhpg, which in turn reduces the dimensionality of the compounds from a 2D rectangular grid network to a 0D molecule structure. Therefore, the formation of compounds with different structures is well-controlled by modulating N-donor ligands, which may provide us with a simple and efficient synthetic route for the tunable construction of compounds.
Scheme 1
 Scheme 1. Observed coordination mode of the ligands: (a) D-Hhpg ligand for compound 1-D; (b) L-Hhpg ligand for compound 1-L; (c) 4,4′-bipy ligand for compounds 1-D and 1-L; (d) D-Hhpg ligand for compound 2-D; (e) 5,5′-BM-2,2′-bipy ligand for compound 2-D
Scheme 1. Observed coordination mode of the ligands: (a) D-Hhpg ligand for compound 1-D; (b) L-Hhpg ligand for compound 1-L; (c) 4,4′-bipy ligand for compounds 1-D and 1-L; (d) D-Hhpg ligand for compound 2-D; (e) 5,5′-BM-2,2′-bipy ligand for compound 2-D2.2 Solid-state CD spectra
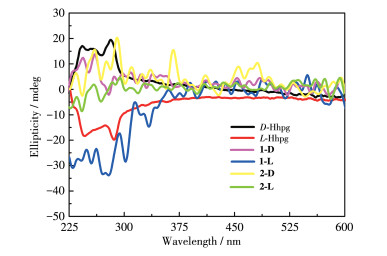
To further demonstrate the homochirality of compounds 1 and 2, solid-state CD analysis has been conducted. As shown in Fig. 8, the CD curve of D-Hhpg displayed a positive Cotton effect with peaks at 249 and 271 nm, while the L-Hhpg displayed a negative Cotton effect at 248 and 272 nm. Compound 1-D displayed positive peak values at 253 and 262 nm. Compound 1-L displayed negative peak values at 252 and 263 nm. Compound 2-D displayed positive peak values at 260 and 290 nm. Compound 2-L displayed negative peak values at 261 and 288 nm. Compounds 1-D/1-L (2-D/2-L) had the opposite CD signal in the same peak position. The result confirms that the 1-D/1-L and 2-D/2-L are homochiral compounds.
Figure 8
2.3 Thermal analyses
The thermal behaviors of compounds 1 and 2 were investigated under a dry nitrogen atmosphere from 20 to 800 ℃ and the TG curves are presented in Fig.S2. 1-D and 1-L had similar thermochemical behaviors, and two weight-loss steps were observed in Fig.S2a and S2b, due to the continuous loss of crystal water, 4,4′-bipy as well as the collapse of crystal structure, respectively. The results show that the main structure of the compounds 1-D/1-L was stable up to 139 ℃/152 ℃. For 2-D (2-L), the first weight loss of 10.16% (11.17%) happened at 31.0-151.0 ℃ (31.0-154.0 ℃), which corresponds to the removal of four lattice water molecules per formula unit (Calcd. 10.55%). From 151.0 to 295.0 ℃ (from 154.0 to 284.0 ℃), the second weight loss of 19.80% (18.39%) corresponds to one and a half lattice water molecules and part 5,5′-BM-2,2′-bipy molecule. Beyond 295.0 ℃ (284.0 ℃), a rapid weight loss was observed, indicating the rapid decomposition of compound 2-D (2-L). Compounds 1 and 2 continued to lose weight up to 800 ℃, indicating the continuous expulsion of organic moieties even at the upper limit of the measurement range.
2.4 PXRD analyses and IR spectra
To check the phase purity of the crystals, the PXRD patterns for compounds 1 and 2 were presented in Fig.S3. The main peaks of simulated patterns of 1-D, 1-L, 2-D, and 2-L match well with their experimental patterns, demonstrating the crystallization degree and the purity of the crystalline phase both are good.
From the IR spectra of compounds 1 and 2 illustrated in Fig.S4, we found that the characteristic peaks of enantiomer compounds were almost completely identical. The νas(COO-) and νs(COO-) vibrations of D-hpg-/L-hpg- could be observed at 1 601-1 655 cm-1 and 1 410-1 513 cm-1, respectively. The peak at 1 224-1 251 cm-1 is due to ν(C—O) vibrations of the phenolic hydroxyl group, and the peak at 1 309-1 378 cm-1 is due to νas(C—N) vibrations of amino groups which connect to the carbon atoms in amino-acids. Moreover, the ν(C—C) and ν(C=N) bending vibration of the pyridine ring at 1 517 and 1 311 cm-1, indicates that the 4,4′-bipy molecules have coordinated with metal in compounds 1-D and 1-L[36]. The ν(C—C) and ν(C=N) bending vibration of the pyridine ring at 1 481-1 485 cm-1 and 1 311-1 323 cm-1 indicates that the 5,5′-BM-2,2′-bipy molecules have coordinated with metal in compounds 2-D and 2-L[37].
2.5 Electrochemical properties of compounds 1 and 2
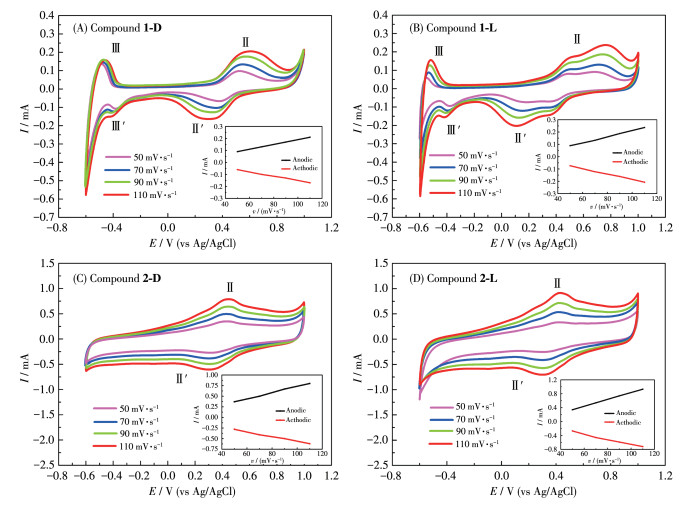
Electrochemical redox properties of compounds 1 and 2 were studied by using CV methods in dilute H2SO4 solution via a three-electrode system. To promote the electroconductivity of the modified electrode, carbon black was employed. The selected mass ratio was 3∶1 for carbon black and solid crystal, in which the largest electrochemical area and smallest impedance were found (Fig.S5). As shown in Fig. 9A and 9B, two pairs of reversible redox peaks (labeled as Ⅱ-Ⅱ′, Ⅲ-Ⅲ′) appeared in the range of the tested potential. The peak potentials of Ⅱ-Ⅱ′ and Ⅲ-Ⅲ′ were 0.551 V(Ⅱ)/0.363 V(Ⅱ′) and -0.468 V(Ⅲ)/-0.403 V(Ⅲ′) for GCE-1-D, and 0.476 V(Ⅱ)/0.337 V(Ⅱ′), -0.512 V(Ⅲ)/-0.382 V(Ⅲ′) for GCE-1-L, respectively. A pair of reversible redox peaks (labeled Ⅱ-Ⅱ′) can be observed in Fig. 9C and 9D. The peak potentials of Ⅱ-Ⅱ′ are 0.449 V(Ⅱ)/0.335 V(Ⅱ′) for GCE-2-D, and 0.433 V(Ⅱ)/0.307 V(Ⅱ′) for GCE-2-L, respectively. The redox peak (Ⅱ-Ⅱ′) should be attributed to the redox process of Co(Ⅱ)/Co(Ⅲ), and the redox peak (Ⅲ-Ⅲ′) should be attributed to the redox process of Co(Ⅱ)/Co(0). The observed irreversibility of these reductions in GCE-1-L is proposed to be due to the decomposition of Co(0) to elemental Co, whose deposition on the working electrode may lead to the observed non-Faradaic current[38]. Moreover, the redox peak currents of compounds 1 and 2 increased as the scan rate increased, and it was found that there was a positive correlation between peak currents and sweep speeds, indicating the surface-controlled feature of the redox process. It is noteworthy that the peak position remained unchanged with the increase in scan speed, indicating the excellent and stable electrochemical redox properties of compounds 1 and 2.
Figure 9
 Figure 9. curves of compounds 1 and 2 with different scan rates in 0.5 mol·L-1 H2SO4 solution
Figure 9. curves of compounds 1 and 2 with different scan rates in 0.5 mol·L-1 H2SO4 solutionInset: plot of the peak current of peak Ⅱ-Ⅱ′ vs scan rate.
2.6 Electrochemical chiral recognition of His enantiomers via CV technique
The interaction between His enantiomers and the chiral interface was measured in 1.0 mmol·L-1 L-His or D-His (40 mL, 0.5 mol·L-1 Na2SO4) solution via CV technique at the potential range from -1 to 1.2 V. As shown in Fig. 10, the bare GCE behaved low and overlapped currents for L-His and D-His, which means no ability of chiral recognition by bare GCE. Significantly increased peak currents of His enantiomers were obtained on GCE-1/2 due to the excellent electrocatalytic properties of compounds 1 and 2. However, the simultaneous shift of peak potential on the other working electrodes resulted in little potential difference for the two enantiomers, indicating hardly to meet the requirement for dual-enantiorecognition. Compared with other working electrodes, the larger peak electric potential difference to L-His and D-His was acquired on GCE-1-D, revealing that 1-D can provide a delicate chiral environment in the enantioselective interaction. These suggest that the chiral compound 1-D has outstanding conductivity and different electrocatalytic functions on the oxidation of D-/L-His. Based on the facts, the strategy for enantiodiscrimination of D-/L-His enantiomers with the chiral compound 1-D material can be achieved.
Figure 10
 Figure 10. CV curves of different modified electrodes for the detection of 1.0 mmol·L-1 D-/L-His in 0.5 mol·L-1 Na2SO4
Figure 10. CV curves of different modified electrodes for the detection of 1.0 mmol·L-1 D-/L-His in 0.5 mol·L-1 Na2SO42.7 Optimization of the experimental condition
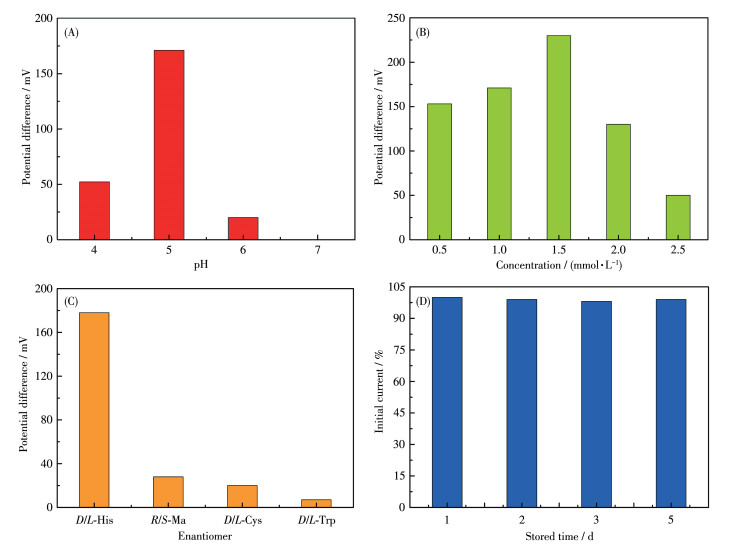
To explore the optimal condition for the enantioselective efficiency of GCE-1-D, we have studied the influence of pH on the peak current ratio between L-His and D-His (Fig.S6). As shown in Fig. 11A, we can easily find that the chiral recognition efficiency (difference of peak potential) gradually increased with the pH ranging from 4.0 to 5.0, and the maximum efficiency at pH=5.0. These anodic peak potentials of L-His and D-His at pH=6.0 or pH=7.0 were not suitable for assaying the enantiomers owing to their weak peak currents. Therefore, pH=5 was chosen as the optimum pH for further chiral recognition measurements.
Figure 11
 Figure 11. (A) Recognition efficiency of GCE-1-D for His isomers in different pH values via CV technique; (B) Recognition efficiency of GCE-1-D in different concentrations of His isomers; (C) Enantioselective interaction with the His, Cys, Ma, and His on GCE-1-D via CV technique; (D) Stability of the GCE-1-D electrode for L-His in different stored times
Figure 11. (A) Recognition efficiency of GCE-1-D for His isomers in different pH values via CV technique; (B) Recognition efficiency of GCE-1-D in different concentrations of His isomers; (C) Enantioselective interaction with the His, Cys, Ma, and His on GCE-1-D via CV technique; (D) Stability of the GCE-1-D electrode for L-His in different stored timesUnder optimal pH of 5, a series of His enantiomers with different concentrations were investigated using the GCE-1-D (Fig.S7a-S7e). Fig. 11B showed that the chiral recognition efficiency (ΔEp=Ep, D-Ep, L) of GCE gradually increased with the increasing concentration of L-His and D-His. Increasing concentration of His isomers from 0.5 to 1.5 mmol·L-1, the chiral recognition efficiency gradually increased and reached the maximum value at 1.5 mmol·L-1, which may be attributed to a state of exactly saturated adsorption, and more than 1.5 mmol·L-1 His isomers might cause the decreasing conductivity of the electrolyte solution. Therefore, 1.5 mmol·L-1 was the optimal concentration of His isomers in this chiral recognition experiment. In addition, the peak potential of GCE-1-D on D-His was greater than that on L-His in this concentration range (Fig.S7f), indicating that the chiral compound-modified electrodes can selectively interact with L-His and D-His.
2.8 Enantioselectivity and stability of the chiral interface
Amino acids enantiomers of cysteine (Cys), tryptophan (Trp), and mandelic acid (Ma) were respectively used to investigate the chiral specificity of the chiral interface (Fig.S8). The recognition efficiency of His was greatly higher than the other three amino acids enantiomers, hinting that this chiral interface possessed high enantioselectivity for His enantiomers (Fig. 11C). Moreover, the peak currents decreased less than 2% compared to the initial response of L-His after the modified electrodes were stored for 5 d at 4 ℃ (Fig. 11D). These results demonstrate that this method possesses acceptable enantioselectivity and stability.
2.9 Electrochemical chiral recognition of His enantiomers via DPV technique
DPV is a powerful method in electrochemical analysis because of its higher sensitivity. Therefore, to further confirm the ability of compound 1-D to recognize His enantiomers, DPV was used to verify its electrochemical recognition performance. Using the same electrode and equipment as the CV technology in the electrolyte solution (40 mL, 0.5 mol·L-1 Na2SO4), the DPV curves for identifying His enantiomers under different pH values were recorded at a scan rate of 50 mV·s-1. The maximum chiral recognition ability was obtained at pH=5 as shown in Fig. 12A. The recognition results from DPVs were consistent with the conclusions of CVs, which demonstrates that the electrode modified by compound 1-D has better electrochemical recognition performance. And under the optimal pH condition, we continue to explore the influence of different concentrations on the recognition efficiency (Fig.S9 and S10). The modified electrode displayed the best recognition effect on 1.0 mmol·L-1 His enantiomers. The electrochemical test results show that the electrode modified by compound 1-D can be used for the recognition of His enantiomers as a promising chiral material to construct electrochemical sensing platforms.
Figure 12
 Figure 12. (A) Recognition efficiency (IL/ID) of GCE-1-D for His isomers in different pH values via DPV technique; (B) Recognition efficiency of GCE-1-D in different concentrations of His isomers via DPV technique
Figure 12. (A) Recognition efficiency (IL/ID) of GCE-1-D for His isomers in different pH values via DPV technique; (B) Recognition efficiency of GCE-1-D in different concentrations of His isomers via DPV technique2.10 Identification of D-/L-His in racemic mixture
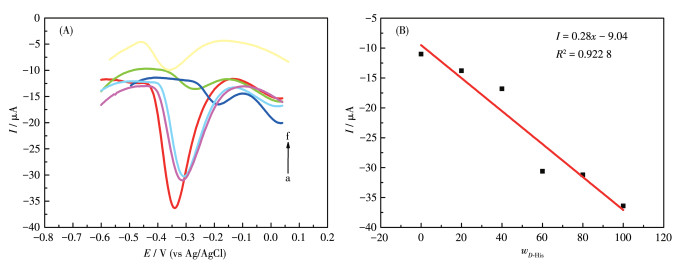
The performance of the chiral sensor to identify the ratio of L-/D-His enantiomers in the racemic solution is significant for further practical applications. The proposed GCE-1-D electrochemical sensor was applied to determine the precise mass fraction of D-His (wD-His) in a racemic mixture with 1 mol·L-1 total concentration. As shown in Fig. 13A, the oxidation peaks of D-His at about 13.5 mV and L-His at about 44.5 mV (Fig.S9b) were combined into a single one, which gradually shifted in the positive direction as D-His concentration increased. More importantly, the oxidation currents against wD-His in the mixture of His enantiomers exhibited a linear dependence as wD-His increased with the correlation coefficient of 0.922 8 (Fig. 13B). These results imply that the GCE-1-D chiral sensor can effectively quantify the ratio of His enantiomers for a racemic sample from a practical point of view. From the perspective of practical application, this has a certain guiding significance for the accurate determination of the drug isomers in a racemic solution.
Figure 13
 Figure 13. (A) DPV curves of GCE-1-D for obtaining wD-His in the mixture of His enantiomers; (B) Linear relationship between wD-His and the oxidation currents
Figure 13. (A) DPV curves of GCE-1-D for obtaining wD-His in the mixture of His enantiomers; (B) Linear relationship between wD-His and the oxidation currentsFrom a to f, the D-His concentratin increased.
3. Conclusions
By using chiral amino acid derivatives D-/L-Hhpg and metal Co ion in the presence of N-donor ancillary ligands, two pairs of enantiomeric coordination compounds have been successfully prepared. Compounds 1-D and 1-L crystallize in the chiral monoclinic space group P21 and show 2D (4, 4) rectangular grid networks that consist of left- or right-handed helical chains. Compound 2-D belongs to the P21 space group of the chiral monoclinic system and exhibits 0D molecule structures. Compounds 2-D and 2-L are enantiomers. Furthermore, compound 1-D can serve as an electrochemical sensor for efficiently detecting His enantiomers and quantitatively determining the enantiomeric excess in the His mixture on account of its electrochemically reversible redox behavior. This work provides an economically cheap and easy-to-handle route for selective identification of chiral His enantiomers.
Acknowledgments: This work was supported by the Natural Science Foundation of Hebei Province (Grant No.B2018205152), the Project of Introduction of Overseas Returnee in Hebei Province (Grant No.CL201715), and the Key Project of the Natural Science Foundation of Hebei Normal University (Grant No.L2021Z03). Supporting information is available athttp://www.wjhxxb.cn
-
-
[1]
Zhao Q Q, Yang J P, Zhang J, Wu D T, Tao Y X, Kong Y. Single-template molecularly imprinted chiral sensor for simultaneous recognition of alanine and tyrosine enantiomers[J]. Anal. Chem., 2019, 91(19): 12546-12552. doi: 10.1021/acs.analchem.9b03426
-
[2]
Chen Z, Wang Q, Wu X, Li Z, Jiang Y B. Optical chirality sensing using macrocycles, synthetic and supramolecular oligomers/polymers, and nanoparticle based sensors[J]. Chem. Soc. Rev., 2015, 44(13): 4249-4263. doi: 10.1039/C4CS00531G
-
[3]
Zor E, Morales-Narváez E, Alpaydin S, Bingol H, Ersoz M, Merkoçi A. Graphene-based hybrid for enantioselective sensing applications[J]. Biosens. Bioelectron., 2017, 87: 410-416. doi: 10.1016/j.bios.2016.08.074
-
[4]
Guo L L, Bao L P, Yang B Z, Tao Y X, Mao H H, Kong Y. Electrochemical recognition of tryptophan enantiomers using self-assembled diphenylalanine structures induced by graphene quantum dots, chitosan and CTAB[J]. Electrochem. Commun., 2017, 83: 61-66. doi: 10.1016/j.elecom.2017.08.024
-
[5]
Chang L M, An Y Y, Li Q H, Gu Z G, Han Y F, Zhan J. N-heterocyclic carbene as a surface platform for assembly of homochiral metal-organic framework thin films in chiral sensing[J]. ACS Appl. Mater. Interfaces, 2020, 12(34): 38357-38364. doi: 10.1021/acsami.0c09578
-
[6]
Li Z Y, Xu H, Wu D T, Zhang J, Liu X R, Gao S M, Kong Y. Electrochemical chiral recognition of tryptophan isomers based on nonionic surfactant-assisted molecular imprinting sol-gel silica[J]. ACS Appl. Mater. Interfaces, 2019, 11(3): 2840-2848. doi: 10.1021/acsami.8b19399
-
[7]
Xu X M, Zhou X M, Qu L, Wang L, Song J T, Wu D H, Zhou W L, Zhou X G, Xiang H F, Wang J, Liu J. Reversible chromatic change of supramolecular gels for visual and selective chiral recognition of histidine[J]. ACS Appl. Bio Mater., 2020, 3(10): 7236-7242. doi: 10.1021/acsabm.0c01063
-
[8]
Zhou Y Y, Xie K, Kong L Y, Chen F, Sun D Y. Highly selective electrochemiluminescent probe to histidine[J]. J. Electroanal. Chem., 2017, 799: 122-125. doi: 10.1016/j.jelechem.2017.05.054
-
[9]
Kopple J D, Swendseid M E. Evidence that histidine is an essential amino acid in normal and chronically uremic man[J]. J. Clin. Investig., 1975, 55(5): 881-891. doi: 10.1172/JCI108016
-
[10]
Kumari B, Kundu S, Ghosh K, Banerjee M, Pradhan S K, Islam S M, Brandão P, Félix V, Das D. Exploring (bio)catalytic activities of structurally characterized Cu(Ⅱ) and Mn(Ⅲ) compounds: Histidine recognition and photocatalytic application of Cu(Ⅱ) complex and derived CuO nano-cubes[J]. Dalton Trans., 2018, 47: 14008-14016. doi: 10.1039/C8DT03007C
-
[11]
Patel G, Menon S. Recognition of lysine, arginine and histidine by novel p-sulfonatocalix[4]arene thiol functionalized gold nanoparticles in aqueous solution[J]. Chem. Commun., 2009, (24): 3563-3565. doi: 10.1039/b905141d
-
[12]
Wang X J, Miao Q Q, Song T G, Yuan Q P, Gao J H, Liang G L. A fluorescent switch for sequentially and selectively sensing copper(Ⅱ) and L-histidine in vitro and in living cells[J]. Analyst, 2014, 139(13): 3360-3364. doi: 10.1039/C4AN00410H
-
[13]
Antoine F R, Wei C I, Littell R C, Marshall M R. HPLC method for analysis of free amino acids in fish using o-phthaldialdehyde precolumn derivatization[J]. J. Agric. Food Chem., 1999, 47(12): 5100-5107. doi: 10.1021/jf990032+
-
[14]
Xia Q, Lin X, Yang C C, Ma J, Fu Y Z. The application of poly(glutathione disulfide)-poly(L-lysine) multilayer films for the enantioselective interaction with ascorbic acid and isoascorbic acid[J]. J. Electrochem. Soc., 2016, 163(14): B744-B750. doi: 10.1149/2.1251614jes
-
[15]
Wu D T, Pan F, Gao L, Tao Y X, Kong Y. Enantioselective limiting transport into a fixed cavity via supramolecular interaction for the chiral electroanalysis of amino acids regardless of electroactive units[J]. Anal. Chem., 2020, 92(20): 13711-13717. doi: 10.1021/acs.analchem.0c00554
-
[16]
Wu D T, Ma C, Pan F, Tao Y X, Kong Y. Strategies to achieve a ferrocene-based polymer with reversible redox activity for chiral electroanalysis of nonelectroactive amino acids[J]. Anal. Chem., 2021, 93(29): 10160-10166. doi: 10.1021/acs.analchem.1c01158
-
[17]
Kuang R, Zheng L Y, Chi Y H, Shi J M, Chen X X, Zhang C C. Highly efficient electrochemical recognition and quantification of amine enantiomers based on a guest-free homochiral MOF[J]. RSC Adv., 2017, 7(19): 11701-11706. doi: 10.1039/C7RA00205J
-
[18]
Dong L Q, Zhang Y S, Duan X M, Zhu X F, Sun H, Xu J K. Chiral PEDOT-based enantioselective electrode modification material for chiral electrochemical sensing: Mechanism and model of chiral recognition[J]. Anal. Chem., 2017, 89(18): 9695-9702. doi: 10.1021/acs.analchem.7b01095
-
[19]
Rodriguez J, Bourissou D. Well-defined chiral gold(Ⅲ) compounds: New opportunities in asymmetric catalysis[J]. Angew. Chem. Int. Ed., 2018, 57(2): 386-388. doi: 10.1002/anie.201710105
-
[20]
He X X, Liu Y, Lv Y, Dong Y Y, Hu G H, Zhou S, Xu Y. L- and D-[LnZn(IN)3(C2H4O2)]n (Ln=Eu, Sm, and Gd): Chiral enantiomerically 3D 3d-4f coordination polymers constructed by interesting butterfly-like building units and —[Ln—O—Zn]n—helices[J]. Inorg. Chem., 2016, 55(5): 2048-2054. doi: 10.1021/acs.inorgchem.5b02372
-
[21]
Wang C H, Kaneti Y V, Bando Y, Lin J J, Liu C, Li J S, Yamauchi Y. Metal-organic framework-derived one-dimensional porous or hollow carbon-based nanofibers for energy storage and conversion[J]. Mater. Horiz., 2018, 5(3): 394-407. doi: 10.1039/C8MH00133B
-
[22]
Cruz C, Gonzalez C, Rubio F, Erices J, Wrighton-Araneda K, Cortés-Arriagada D, Venegas-Yazigi D, Audebrand N, Paredes-García V. Chiral 1D metal-organic materials based on Cu(Ⅱ) and amino acid Schiff bases[J]. Cryst. Growth Des., 2021, 22(1): 237-250.
-
[23]
Zilberg R A, Berestova T V, Gizatov R R, Teres Y B, Galimov M N, Bulysheva E O. Chiral selectors in voltametric sensors based on mixed phenylalanine/alanine Cu(Ⅱ) and Zn(Ⅱ) complexes[J]. Inorganics, 2022, 10(8): 112. doi: 10.3390/inorganics10080112
-
[24]
Zhang X, Wang J, Yang S D. Enantioselective cobalt-catalyzed reductive cross-coupling for the synthesis of axially chiral phosphine-olefin ligands[J]. ACS Catal., 2021, 11(22): 14008-14015. doi: 10.1021/acscatal.1c04128
-
[25]
Areas E S, Junior H C, Freitas B P, Ferreira G B, Guedes G P. Homobinuclear compounds based on a chiral oxazolidine ligand: from solid state study to aqueous solution dynamics[J]. Inorg. Chim. Acta, 2022, 529: 120664. doi: 10.1016/j.ica.2021.120664
-
[26]
Zhai B, Li Z Y, Zhang X F, Wu X X, Guo J H, Huo J Z, Ding B. Synthesis, structures, magnetic and luminescent properties of a series of iron(Ⅱ) and zinc(Ⅱ) coordination frameworks with versatile 4-substituted 1,2,4-triazole ligands[J]. Z. Anorg. Allg. Chem., 2016, 642(3): 260-267. doi: 10.1002/zaac.201500768
-
[27]
Wang Y L, Chen L, Liu C M, Du Z Y, Chen L L, Liu Q Y. 3D chiral and 2D achiral cobalt(Ⅱ) compounds constructed from a 4-(benzimidazole-1-yl) benzoic ligand exhibiting field-induced single-ion-magnet-type slow magnetic relaxation[J]. Dalton Trans., 2016, 45(18): 7768-7775. doi: 10.1039/C6DT00676K
-
[28]
Zhang T, Huang H Q, Cheng X Y, Guo D, Mei H X, Huang R B, Zheng L S. The synthesis and chiral crystal structures of two enantiomers of a Ag helical coordination polymer based on argentophilicity[J]. CrystEngComm, 2016, 18: 670-673. doi: 10.1039/C5CE01948F
-
[29]
Mei H X, Zhang T, Wang D F, Huang R B, Zheng L S. A Zn-oxalate helix linked by a water helix: Spontaneous chiral resolution of a Zn helical coordination polymer[J]. New J. Chem., 2015, 39: 2075-2080. doi: 10.1039/C4NJ02017K
-
[30]
Durá G, Carrión M C, Jalón F A, Rodríguez A M, Manzano B R. Self-assembly of silver(Ⅰ) and ditopic heteroscorpionate ligands. Spontaneous chiral resolution in helices and sequence isomerism in coordination polymers[J]. Cryst. Growth Des., 2013, 13: 3275-3282. doi: 10.1021/cg400636a
-
[31]
Ou G C, Li Z Z, Zhang M, Yuan X Y. Chiral resolution of L- and D-alanine and a racemic macrocyclic nickel(Ⅱ) complex: Synthesis and crystal structures[J]. Transition Met. Chem., 2014, 39: 135-140. doi: 10.1007/s11243-013-9782-9
-
[32]
Tao W A, Zhang D X, Nikolaev E N, Cooks R G. Copper(Ⅱ)-assisted enantiomeric analysis of D, L-amino acids using the kinetic method: Chiral recognition and quantification in the gas phase[J]. J. Am. Chem. Soc., 2000, 122: 10598-10609. doi: 10.1021/ja000127o
-
[33]
Sheldrick G M. SHELXS-97, Program for the solution of crystal structures. University of Göttingen, Germany, 1997.
-
[34]
Sheldrick G M. SHELXL-97, Program for the refinement of crystal structures. University of Göttingen, Germany, 1997.
-
[35]
Wei H W, Yang Q F, Lai X Y, Wang X Z, Yang T L, Hou Q, Liu X Y. Field-induced slow relaxation of magnetization in a distorted octahedral mononuclear high-spin Co(Ⅱ) complex[J]. CrystEngComm, 2018, 20(7): 962-968. doi: 10.1039/C7CE01981E
-
[36]
Mikuriya M, Indrawati R, Hashido R, Matsubara S, Nakamura C, Yoshioka D, Yokota K, Fukuzaki M, Handa M. Chain compounds based on paddle-wheel copper(Ⅱ) carboxylate bearing four nitroxide radicals[J]. Magnetochemistry, 2018, 4(2): 22. doi: 10.3390/magnetochemistry4020022
-
[37]
Jassal A K. Advances in ligand-unsupported argentophilic interactions in crystal engineering: An emerging platform for supramolecular architectures[J]. Inorg. Chem. Front., 2020, 7(19): 3735-3764. doi: 10.1039/D0QI00447B
-
[38]
Liang K L, Lu L J, Liu X, Yang D L, Wang S C, Gao Y M, Alhumade H, Yi H, Lei A W. Electrochemical cobalt-catalyzed cyclotrimerization of alkynes to 1,2,4-substituted arenes[J]. ACS Catal., 2021, 11(24): 14892-14897. doi: 10.1021/acscatal.1c04639
-
[1]
-
Figure 1 Coordination environment of Co(Ⅱ) in compounds 1-D (A) and 1-L (B)
H atoms have been omitted for clarity.
Figure 2 (A) One-dimensional right-handed helical chains of compound 1-D; (B) 1D left-handed helical chains of compound 1-L
Figure 5 View of coordination environment of Co(Ⅱ) in compound 2-D
H atoms are omitted for clarity.
Scheme 1 Observed coordination mode of the ligands: (a) D-Hhpg ligand for compound 1-D; (b) L-Hhpg ligand for compound 1-L; (c) 4,4′-bipy ligand for compounds 1-D and 1-L; (d) D-Hhpg ligand for compound 2-D; (e) 5,5′-BM-2,2′-bipy ligand for compound 2-D
Figure 9 curves of compounds 1 and 2 with different scan rates in 0.5 mol·L-1 H2SO4 solution
Inset: plot of the peak current of peak Ⅱ-Ⅱ′ vs scan rate.
Figure 10 CV curves of different modified electrodes for the detection of 1.0 mmol·L-1 D-/L-His in 0.5 mol·L-1 Na2SO4
Figure 11 (A) Recognition efficiency of GCE-1-D for His isomers in different pH values via CV technique; (B) Recognition efficiency of GCE-1-D in different concentrations of His isomers; (C) Enantioselective interaction with the His, Cys, Ma, and His on GCE-1-D via CV technique; (D) Stability of the GCE-1-D electrode for L-His in different stored times
Figure 12 (A) Recognition efficiency (IL/ID) of GCE-1-D for His isomers in different pH values via DPV technique; (B) Recognition efficiency of GCE-1-D in different concentrations of His isomers via DPV technique
Figure 13 (A) DPV curves of GCE-1-D for obtaining wD-His in the mixture of His enantiomers; (B) Linear relationship between wD-His and the oxidation currents
From a to f, the D-His concentratin increased.
Table 1. Crystal data and structure refinements of the compounds
Parameter 1-D 1-L 2-D Empirical formula C18H20N3ClCoO5 C18H20N3ClCoO5 C28H39ClCoN4O11.5 T/K 298(2) 298(2) 298(2) Formula weight 452.75 452.75 709.92 Space group P21 P21 P21 Crystal system Monoclinic Monoclinic Monoclinic a/nm 1.022 64(13) 1.011 47(9) 1.083 99(11) b/nm 0.909 25(11) 0.902 04(8) 1.353 46(14) c/nm 1.153 49(15) 1.142 67(11) 2.186 3(2) β/(°) 111.806(4) 111.754(4) 91.775(3) V/nm3 0.995 8(2) 0.968 31(16) 3.206 1(6) Z 2 2 4 Dc/(Mg·m-3) 1.510 1.553 1.266 F(000) 466 466 1 264 θ range/(°) 1.902-25.009 2.168-25.016 2.07-29.13 Reflection collected 6 348 5 108 54 80 Independent reflection 2 728 (Rint=0.071 4) 3 043 (Rint=0.052 5) 4 792 (Rint=0.053 2) Data, restraint, number of parameters 2 728, 265, 206 3 043, 1, 212 14 629, 241, 725 Goodness-of-fit on F2 1.236 1.092 1.032 R1, wR2 [I > 2σ(I)] 0.102 1, 0.227 0 0.094 4, 0.203 3 0.070 0, 0.126 9 R1, wR2 (all data) 0.143 5, 0.248 2 0.139 1, 0.224 1 0.129 5, 0.143 1 Flack parameter -0.06(7) 0.05(7) 0.00(3)  下载: 导出CSV
下载: 导出CSV
-


















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 2
- 文章访问数: 2263
- HTML全文浏览量: 162
 下载:
下载:
