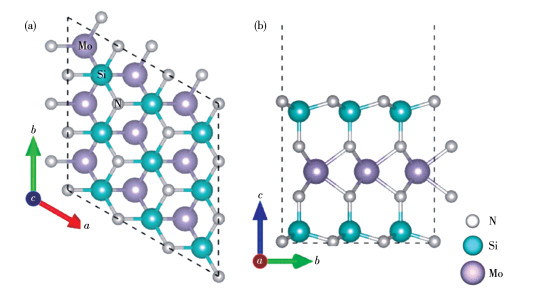
图 1.
单层MoSi2N4的3×3×1超胞俯视图(a)和侧视图(b)
Figure 1.
Top view (a) and side view (b) of MoSi2N4 monolayer with 3×3×1 supercell

锂离子电池是一种二次电池,其主要依靠锂离子在正负极之间移动来工作,具有较高的比容量和工作电压、无记忆效应及较长的循环寿命,被广泛运用于可移动储能等领域[1-2]。随着现代社会对储能器件性能需求的增加,以往锂离子电池的石墨电极已不能满足人们的需求。由于整个二维材料家族均有利于电解质渗透和界面反应的多孔结构及较大的比表面积,可提供更多的锂离子储存活性位点与通道,因此其在电极材料应用方面有着巨大潜力[3-5]。
相关研究表明,最初的二维材料石墨烯具有540 mAh·g-1的储锂容量[6],之后经合成工艺的改进后得到的花状石墨烯的储锂容量达到了640 mAh·g-1 [7],比以往的石墨材料储锂容量(372 mAh·g-1)增加了72%。在理论研究方面,学者们对锂离子在石墨烯表面的吸附性质[8-10]及扩散行为[11-13]也有了一定认识,发现硼、氮掺杂石墨烯可提升其锂离子存储与扩散能力[14-15]。但无论是合成工艺的改进或是掺杂改性,均不能避免石墨烯作为电极材料时因大电流产生的极化,同时石墨烯表面也易受化学状态影响,在其用作电极材料时欠缺安全性。因此,人们将目光移向二硫化钼,研究了其溶剂化锂离子共嵌入的特性,以此来阻止固体电解质界面膜的形成,此时二硫化钼表现出273.5 mAh·g-1的储锂容量[16]。之后人们将二硫化钼天然矿石作为锂离子电池负极材料时表现出1 199 mAh·g-1的储锂容量[17],高于石墨烯。在研究锂离子在二硫化钼表面的吸附及扩散行为时,人们发现锂离子在二硫化钼表面扩散时所需克服的势垒大于常温下热运动的能量级别,锂离子扩散过程不能自发进行[18],导致二硫化钼在用作电极材料时倍率不佳。因此,寻找一种同时兼顾高倍率及稳定性的新型电极材料至关重要。
最近,研究人员发现了一种新型二维层状材料MoSi2N4,其具有1.94 eV的半导体带隙,优于二硫化钼的理论载流子迁移率,具有约66 GPa的高强度及出色的环境稳定性等优点[19],被认为是一种潜在的锂离子电池电极材料。目前大量的研究集中在MoSi2N4的压电性及光催化方面[20],而MoSi2N4的电化学储锂机制是类似于CoNiO2[21]结构中的锂离子转换反应还是类似于Li5Cr7Ti6O25[22]上的锂离子嵌入反应尚不明确,因此急需对其储锂能力及储锂机制进行系统地研究。并且由于其结构的特殊性,很难通过实验直接探测电荷分布、离子储存及扩散等微观数据,而采用理论计算的方法可以获得实验难以获取的有用信息。综上所述,我们决定基于密度泛函理论的第一性原理,在单层MoSi2N4超胞内掺杂单个硼原子改进其锂存储性能,并探究其储锂机制,为MoSi2N4作为锂离子电池电极材料的应用提供理论指导。
采用基于密度泛函理论的平面波超软赝势方法研究了硼原子掺杂对MoSi N锂离子存储及扩散性能的影响。采用的交换关联势为广义梯度近似(GGA)[23],价电子与离子实之间波函数的连续化处理采用RPBE[24]赝势描述。在描述原子实与价电子之间的相互作用时,选取的价电子组态分别为Li:2s1、B:2s22p1、N:2s22p3、Si:3s23p2、Mo:4s24p64d55s1。平面波截断能设置为330 eV,布里渊区k 点网格均选取3×3×1[25]。结构优化选用的自恰收敛精度设置为5×10-5 eV·atom-1,原子间的力场收敛精度设置为1 eV·nm-1,最大应力设置为0.2 GPa,最大原子位移设置为5×10-4 nm。为减少MoSi N层间范德华作用,将单层MoSi2N4的真空层设置为1.5 nm。全部计算过程均在CASTEP软件包下完成。
二维层状材料MoSi2N4是一种空间群为P63/mmc 的六方晶体,单层包含N-Si-N-Mo-N-Si-N七个原子层。在3×3×1超胞中含有63个原子,其中钼原子为9个,硅原子为18个,氮原子为36个。所有的硅原子周围的3个氮原子形成三配位,所有的钼原子与周围的氮原子形成六配位,其结构如图 1所示。对MoSi2N4的3×3×1超胞进行几何优化,得到晶格常数a=b=0.872 nm,其与实验上单层MoSi2N4的晶格常数差值不超过1%(MoSi2N4:a=b=0.882 nm)[19]。
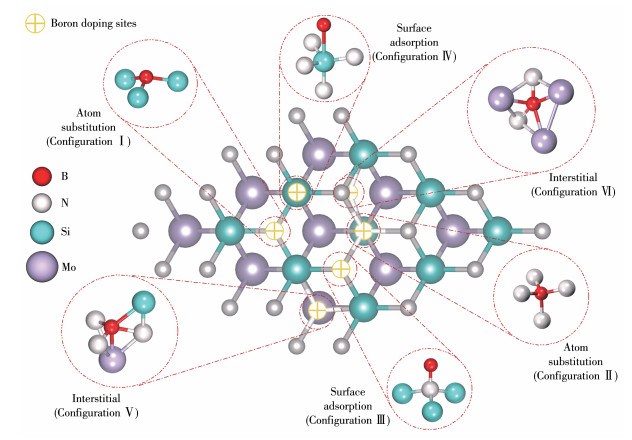
考虑到单层MoSi2N4表面无缺陷,其对锂离子作用力相对较弱,需通过掺杂改性的方式,使其整体呈电子缺失状态,以增强MoSi2N4对锂离子的吸附作用。尺度为3×3×1的单层MoSi2N4超胞已符合掺杂与吸附的研究条件,可类比钴掺杂MoS2的析氢及析氧性能的研究[26]。在建立硼掺杂MoSi2N4的物理模型时,均以单个硼原子掺杂,并考虑了3类掺杂模式,分别为原子替代、表面吸附及间隙掺杂。根据如图 2中的掺杂位点,建立了6种硼掺杂物理模型。其中,原子替代模型为硼替代氮位(构型Ⅰ)与硼替代硅位(构型Ⅱ),表面吸附模型为硼吸附在氮顶位(构型Ⅲ)与硼吸附在硅顶位(构型Ⅳ),间隙掺杂模型为硼在钼上方间隙(构型Ⅴ)与硼在氮下方间隙(构型Ⅵ)。特别地,为保证MoSi2N4的稳定性,结构中心的钼原子不适合作为替换掺杂位点纳入考虑。
不同掺杂源中杂原子所包含的绝对化学势差异较大,需要通过掺杂形成能的计算来筛选有效的掺杂源及稳定的掺杂构型。2类掺杂形成能公式:
|
|
(1) |
|
|
(2) |
其中Esub为替换位掺杂形成能,Ead为吸附与间隙掺杂形成能,Eb为硼掺杂MoSi2N4超胞总能量,Ea为MoSi2N4超胞总能量,μX为被替换原子的化学势,μB为氮原子的化学势。出于对掺杂模型的热力学稳定性考虑,原子化学势的零点一般选取稳定单质晶体中单个原子的能量[27]。因此,计算过程选取了氮原子在1.000 nm3立方真空晶胞中氮气分子的化学势,优化后N≡N键长为0.163 nm,单个氮原子能量为-271.080 eV;选取了硅原子在晶体硅中的化学势,单个硅原子能量为-107.520 eV;选取了硼原子在硼单质中的化学势,单个硼原子能量为-76.536 eV。掺杂形成能越负说明该构型的热力学稳定性越强,计算得到的不同位点掺杂硼原子的形成能结果记录在表 1中。由表可知,硼原子替代表面氮原子的构型Ⅰ最为稳定,因此选取该构型作为锂离子吸附基底进行下一步研究。
 下载:
导出CSV
下载:
导出CSV
| Doped mode | Configuration | Lattice constant | Cell volume / nm3 | Formation energy / eV | ||
| a /nm | b /nm | c /nm | ||||
| Substitution | Ⅰ | 0.876 | 0.876 | 1.640 | 1.258 | -2.02 |
| Ⅱ | 0.870 | 0.870 | 1.940 | 1.468 | -0.40 | |
| Adsorption | Ⅲ | 0.875 | 0.875 | 1.975 | 1.512 | 5.11 |
| Ⅳ | 0.874 | 0.874 | 1.936 | 1.479 | 5.57 | |
| Interstitial | Ⅴ | 0.873 | 0.872 | 1.983 | 1.510 | 4.69 |
| Ⅵ | 0.882 | 0.882 | 1.972 | 1.534 | 5.80 | |
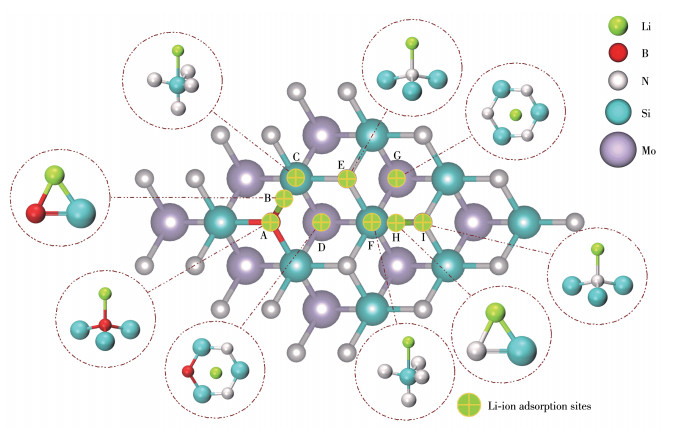
在建立锂离子吸附构型时,以构型Ⅰ吸附单个锂离子。当硼替换表面氮后,MoSi2N4原本的对称性受到破坏,吸附过程受到硼原子电子局域态的影响,在3×3×1的超胞内同原子顶会出现2种不同的吸附情况。依照图 3的吸附位点,建立了9种锂离子吸附的物理模型,分别为锂离子吸附在硼顶位(A)、硅顶位(C和F)及氮顶位(E和I)、硼硅桥位(B)及氮硅桥位(H),由于锂离子的半径较大(RLi+=0.076 nm),间隙位只考虑了锂离子吸附在表层三硅中心间隙(D和G)。
锂离子吸附能力是衡量电极材料储锂能力最重要的方面之一,通过比较各位点锂离子吸附能大小,可得到硼掺杂MoSi2N4最佳的锂离子储存位点。吸附能(Ead′)公式:
|
|
(3) |
其中Eb′为构型Ⅰ的总能量,Ec为锂离子吸附在构型Ⅰ表面的总能量。μLi为锂离子的绝对化学势,通过计算金属锂中单个锂的单点能,可知其为-190.680 eV。吸附能越小说明锂离子储存在该位点的稳定性越强,计算得到的不同位点锂离子吸附能如表 2所示。结果表明,MoSi2N4表层满配位导致其锂离子吸附性能较差,在硼原子替换氮原子后,MoSi2N4表面整体呈电子缺失状态,这使得其锂离子的吸附作用显著增强,这一现象与p型掺杂二硫化钼表面锂离子吸附行为相同[28]。在构型Ⅰ表面,同种吸附位点的锂离子受硼局域态影响越小则吸附能越大,其中氮顶位(E和I)为最佳的锂离子吸附位点。
下载:
导出CSV
| Li-ion adsorption site | Li-ion adsorption energy (before doped) / eV | Li-ion adsorption energy (after doped) / eV | Distance between Li-ion and boron / nm |
| A | 1.19 | -1.54 | 0.226 |
| B | 1.13 | -1.60 | 0.247 |
| C | 1.12 | -1.66 | 0.292 |
| D | 1.26 | -1.78 | 0.261 |
| E | 1.19 | -1.87 | 0.361 |
| F | 1.12 | -1.72 | 0.407 |
| G | 1.26 | -1.88 | 0.483 |
| H | 1.13 | -1.76 | 0.439 |
| I | 1.19 | -1.91 | 0.548 |
将锂离子聚合形成锂单质的能量定义为锂聚合能。为简单了解多数锂离子吸附时的稳定性,比较了锂聚合能与锂离子在构型Ⅰ表面的吸附能。若锂聚合能低于锂吸附能,则锂离子吸附数量增加后会使锂离子在MoSi2N4表面聚合形成锂单质,导致后续的吸附过程受阻;若锂聚合能高于锂吸附能,则吸附过程受锂离子吸附数量影响较小。结果表明,锂聚合能为-1.000 eV,此结果高于所有位点的锂离子吸附能,说明掺杂后锂离子吸附数量的增多不会导致表面形成锂块而降低MoSi2N4锂储存的稳定性。
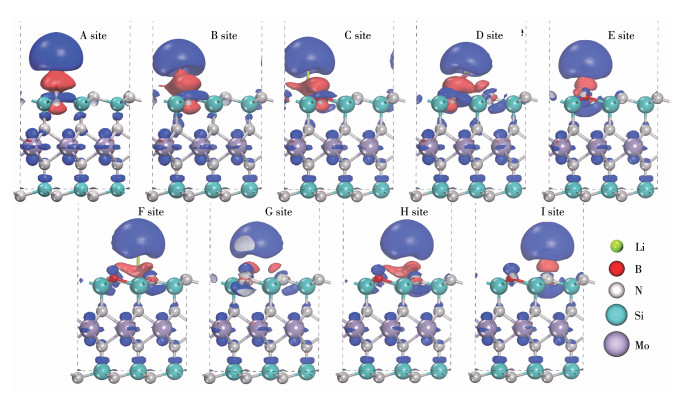
计算得到不同锂离子吸附在构型Ⅰ不同位点下的电子密度差分如图 4所示,其中蓝色区域表示电子密度减少,红色区域表示电子密度增加。锂离子在所有吸附构型中均以电子给体的形式存在。当锂离子吸附在硼顶(A位点)或硼硅桥(B位点)时,硼周围呈现较强的电子局域性。当锂离子吸附在硅顶(C位点)或三硅中心(D位点)时,由于锂离子距离硼较近,硼表面与硼硅桥表面依然显示出较强的电子局域性,而硅表面几乎不储存电子。当锂离子吸附在距离硼较远的硅顶(F位点)或氮硅桥(H位点) 时,多数电子集中在氮硅桥与氮表面,极少量电子以片状形态分布在硅表面,同样体现硅在该体系中较差的电子储存能力。当锂离子吸附在氮顶(E位点)或三硅中心(G位点)时,锂离子提供的电子以球状形态储存在氮的上表面,说明构型Ⅰ表面的氮也具备一定的电子储存能力。
锂离子的扩散性能直接决定了锂离子电池的充放电倍率,而第一性原理无法直接得到其扩散速率,为将计算结果与实验结果相关联,需引入阿伦尼乌斯公式:
|
|
(4) |
其中k为锂离子扩散速率,A为频率因子,Ea为反应活化能,R为摩尔气体常数,T为热力学温度。在本研究中,A、R、T均为定量,反应活化能Ea可近似等于利用过渡态搜索计算出的扩散势垒Edb。据公式4可知,扩散势垒越小,锂离子在体系表面沿某路径扩散的速率越快。因此,我们计算了锂离子在构型Ⅰ表面扩散时的路径及势垒,根据锂离子在其表面的吸附能(表 2)及相应吸附位点(图 3)绘制了简单的势能面,如图 5所示。

锂离子的扩散必然是由吸附能较高的位点向吸附能较低的位点进行,为使这一过程能自发进行,可对I→G→E、I→E、G→D、D→F、D→C、E→D→ A及E→A这7条扩散路径进行过渡态搜索,以扩散路径分数坐标及扩散过程系统整体的能量变化反映锂离子扩散情况,详细情况记录在图 6。锂离子在相邻氮顶位扩散时(I→E)的势垒为0.165 eV,大于其间接向受硼局域态影响较小的三硅中心位扩散再到氮顶位(I→G→E)的势垒(0.085 eV)。当锂离子到达氮顶位时还可能直接向硼顶位扩散(E→A),扩散过程需克服0.122 eV的势垒,该结果小于其途经受硼局域态影响较大的三硅中心位再到硼顶位扩散时(E→D→A)的势垒(0.341 eV)。锂离子在相邻三硅中心位扩散(G→D)的势垒为0.146 eV,并且在扩散至受硼局域态影响较大的三硅中心(D)位时还可能向周围的硅顶位扩散,其扩散向离硼较远的硅顶位(D→F)时需克服0.077 eV的势垒,该结果小于扩散至距离硼较近的硅顶位(D→C)所需克服0.123 eV的势垒。特别地,支持文件(Supporting information) 内锂离子在不同路径扩散的过渡态特征分析也证实了这些结论。
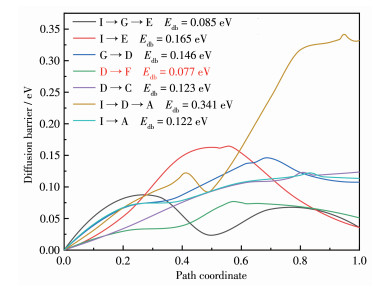
结果表明,锂离子扩散过程总有避开硼局域态影响趋势,这个现象在硼掺杂金刚石中被证实[29]。特别地,锂离子在D→F这一扩散路径下具有最低的势垒(0.077 eV),低于石墨烯及其衍生物(0.191~ 0.385eV)[30]和二硫化钼(0.190eV)[18]上锂离子扩散所需克服的势垒,说明硼掺杂MoSi2N4用作电极材料时,在充放电倍率方面有着更大的优势。
采用平面波超软赝势的方法研究了硼掺杂MoSi2N4锂离子吸附与扩散行为。(1) 通过掺杂形成能的计算,发现硼原子替代MoSi2N4表面氮原子的构型最为稳定。(2) 通过锂离子吸附能的计算,发现硼替代氮后MoSi2N4表面锂离子吸附能增大,并且锂离子吸附位置距硼越远,吸附能越大。(3) 电子密度差分图表明了锂总是呈电子缺失态,硼与氮均会储存锂供给的电子。(4) 根据锂离子吸附在不同位点的吸附势能面得出了7条可能的扩散路径,对这些路径进行过渡态搜索,得出锂离子在硼掺杂MoSi2N4表面有着远离硼扩散的趋势,锂离子在D→F这一路径下扩散需克服0.077 eV的低势垒。硼原子的引入使得锂离子吸附及扩散性能均得到了提高,为MoSi2N4作为锂离子电池电极材料的应用提供了一种理论上可行的方案。
Supporting information is available at http://www.wjhxxb.cn
王鹏博, 郑俊超. 锂离子电池的发展现状及展望[J]. 自然杂志, 2020,39,(4): 283-289. WANG P B, ZHENG J C. The Present Situation and Expectation of Lithium-Ion Battery[J]. Chinese Journal of Nature, 2020, 39(4): 283-289.
周恒辉, 慈云翔, 刘昌炎. 锂离子电池电极材料的研究进展[J]. 化学进展, 1998,10,(1): 85-94. doi: 10.3321/j.issn:1005-281X.1998.01.008ZHOU H H, CI Y X, LIU C Y. Progress in Studies of the Electrode Materials for Li-Ion Batteries[J]. Prog. Chem., 1998, 10(1): 85-94. doi: 10.3321/j.issn:1005-281X.1998.01.008
Novoselov K S, Geim A K. Electric Field Effect in Atomically Thin Carbon Films[J]. Science, 2004, 306(5696): 666-669. doi: 10.1126/science.1102896
Guo P, Song H H. Electrochemical Performance of Graphene Nanosheets as Anode Material for Lithium-Ion Batteries[J]. Electrochem. Commun., 2009, 11(6): 1320-1324. doi: 10.1016/j.elecom.2009.04.036
Pan D Y, Wang S, Zhao B, Wu M H, Zhang H J, Wang Y, Zheng J. Li Storage Properties of Disordered Graphene Nanosheets[J]. Chem. Mater., 2009, 21(14): 3136-3142. doi: 10.1021/cm900395k
Yoo E, Kim J, Hosono E, Zhou H S, Kudo T, Honma I. Large Reversible Li Storage of Graphene Nanosheet Families for Use in Rechargeable Lithium Ion Batteries[J]. Nano Lett., 2008, 8(8): 2277-2282. doi: 10.1021/nl800957b
Cresti A, Carrete J, Okuno H, Wang T, Madsen G K H, Mingo N, Pochet P. Growth, Charge and Thermal Transport of Flowered Graphene[J]. Carbon, 2020, 161: 259-268. doi: 10.1016/j.carbon.2020.01.040
Khantha M, Cordero N A, Molina M L, Alonso J A, Girifalco L A. Interaction of Lithium with Graphene: An Ab Initio Study[J]. Phys. Review. B, 2004, 70(12): 125422. doi: 10.1103/PhysRevB.70.125422
Chan K T, Neaton J B, Cohen M L. First-Principles Study of Metal Adatom Adsorption on Graphene[J]. Phys. Rev. B, 2008, 77(23): 235430. doi: 10.1103/PhysRevB.77.235430
Suzuki T, Hasegawa T, Hajime T M. A Theoretical Study on Storage States of Li-Ions in Carbon Anodes of Li-Ion Batteries Using Molecular Orbital Calculations[J]. Carbon, 2003, 41(10): 1933-1999. doi: 10.1016/S0008-6223(03)00178-7
Toyoura K, Koyama Y, Kuwabara A, Oba F, Tanaka I. First-Principles Approach to Chemical Diffusion of Lithium Atoms in a Graphite Intercalation Compound[J]. Phys. Rev. B, 2008, 78(21): 214303. doi: 10.1103/PhysRevB.78.214303
Ong P S, Chevrier V L, Hautier G, Jain A, Moore C, Kim S, Ma X H, Ceder G. Voltage, Stability and Diffusion Barrier Differences between Sodium-Ion and Lithium-Ion Intercalation Materials[J]. Energy Environ. Sci., 2011, 4(9): 3680-3688. doi: 10.1039/c1ee01782a
Fan X F, Zheng W T, Kuo J L. Adsorption and Diffusion of Li on Pristine and Defective Graphene[J]. ACS Appl. Mater. Interfaces, 2012, 4(5): 2432-2438. doi: 10.1021/am3000962
任王瑜, 侯世成, 姜孝男, 陈卫祥. MoS2/硼掺杂石墨烯的电化学析氢和储锂性能[J]. 浙江大学学报(工学版), 2020,54,(8): 1628-1636. REN W Y, HOU S C, JIANG X N, CHEN W X. MoS2/B-Doped Graphene for Electrochemical Hydrogen Evolution and Lithium Storage[J]. Journal of Zhejiang University (Engineering Science), 2020, 54(8): 1628-1636.
沈进冉, 郭翠静, 陈赫, 周淑琴, 徐斌, 官亦标. 高性能氮掺杂石墨烯的制备及其储锂性能[J]. 储能与科学技术, 2019,8,(6): 1137-1144. SHEN J R, GUO C J, CHEN H, ZHOU S Q, XU B, GUAN Y B. Synthesis and Lithium Storage Property of High-Performance N-Doped Reduced Graphene Oxide[J]. Energy Storage Science and Technology, 2019, 8(6): 1137-1144.
Zhang K, Yoon G, Zhang J, Park M, Kang K, Kang Y M. Pseudocapacitive Behaviour and Ultra-Fast Kinetics from Solvated Ion Cointercalation into MoS 2 for Its Quick Alkali Ion Charging[J]. ACS Appl. Energy Mater., 2019, 2(5): 3726-3735. doi: 10.1021/acsaem.9b00445
Li S J, Tang H H, Ge P, Jiang F, Zhou J H, Zhang C Y, Hou H S, Sun W, Ji X B. Electrochemical Investigation of Natural Ore-Molybdenite (MoS2) as First-Hand Anode for Lithium Storages[J]. ACS Appl. Mater. Interfaces, 2018, 10(7): 6378-6389. doi: 10.1021/acsami.7b18571
张政委, 王强, 薛彩龙, 刘秀萍, 张爱华, 李梦雪. 单层MoS2负极材料储锂行为第一性原理研究[J]. 电子质量, 2020,402,(9): 6-14. doi: 10.3969/j.issn.1003-0107.2020.09.002ZHANG Z W, WANG Q, XUE C L, LIU X P, ZHANG A H, LI M X. First Principles Study on Lithium Storage Behavior of Monolayer MoS 2 Anode Material[J]. Electronics Quality, 2020, 402(9): 6-14. doi: 10.3969/j.issn.1003-0107.2020.09.002
Hong Y L, Liu Z B, Wang L, Zhou T Y, Ma W, Xu C, Shun F, Chen L, Chen M L, Sun D M, Chen X Q, Cheng H M, Ren W C. Chemical Vapor Deposition of Layered Two-Dimensional MoSi2N4 Materials[J]. Science, 2020, 6504(369): 670-674.
Mortazavi B, Javvaji B, Shojaei F, Rabczuk T, Shapeev A V, Zhuang X Y. Exceptional Piezoelectricity, High Thermal Conductivity and Stiffness and Promising Photocatalysis in Two-Dimensional MoSi2N4 Family Confirmed by First-Principles[J]. Nano Energy, 2021, 82: 105716. doi: 10.1016/j.nanoen.2020.105716
Yi T F, Mei J, Peng P P, Luo S H. Facile Synthesis of Polypyrrole-Modified Li5Cr7Ti6O25 with Improved Rate Performance as Negative Electrode Material for Li-Ion Batteries[J]. Composites Part B, 2019, 167: 566-572. doi: 10.1016/j.compositesb.2019.03.032
Yi T F, Qu J P, Lai X, Han X, Chang H, Zhu Y R. Toward High-Performance Li Storage Anodes: Design and Construction of Spherical Carbon-Coated CoNiO2 Materials[J]. Mater. Today Chem., 2021, 19: 100407. doi: 10.1016/j.mtchem.2020.100407
Kohn W, Sham L J. Self-Consistent Equations Including Exchange and Correlation Effects[J]. Phys. Rev., 1965, 140(4A): A1133-A1138. doi: 10.1103/PhysRev.140.A1133
Hammer B, Hansen L B, Norskov J K. Improved Adsorption Energetics within Density-Functional Theory Using Revised Perdew-Burke-Ernzerhof Functionals[J]. Phys. Rev. B, 1999, 59(11): 7413-7421. doi: 10.1103/PhysRevB.59.7413
Monkhorst H J, Pack J D. Special Points for Brillonin-Zone Integrations[J]. Phys. Rev. B, 1976, 13(12): 5188-5192. doi: 10.1103/PhysRevB.13.5188
Xiong Q Z, Wang Y, Liu P F, Zheng L R, Wang G Z, Yang H G, Wong P K, Zhang H, Zhao H J. Cobalt Covalent Doping in MoS2 to Induce Bifunctionality of Overall Water Splitting[J]. Adv. Mater., 2018, 30(29): 180450.
Wang Q, Yu H T, Xie Y, Li M X, Yi T F, Guo C F, Song Q S, Lou M, Fan S S. Structural Stabilities, Surface Morphologies and Electronic Propertiesof Spinel LiTi2O4 as Anode Materials for Lithium-Ion Battery: A First-Principles Investigation[J]. J. Power Sources, 2016, 319: 185-194. doi: 10.1016/j.jpowsour.2016.04.039
刘海俐. 利用第一性原理计算研究低维材料对金属原子的吸附及其在锂离子电池电极材料中的应用. 苏州: 苏州大学, 2019.LIU H L. Investigations on Adorption ofMetalAtom on Low-Dimensional Materials and Its Applications in Li-Ion Batteries by First-Principle Calculations. Suzhou: Soochow University, 2019.
Rehnlund D, Lindgren F, Böhme S, Nordh T, Zou Y, Pettersson J, Bexell U, Boman M, Edström K, Nyholm L. Lithium Trapping in Alloy Forming Electrodes and Current Collectors for Lithium Based Batteries[J]. Energy Environ. Sci., 2017, 10(6): 1350-1357. doi: 10.1039/C7EE00244K
李沛艾. Li在石墨烯、BCx及CxN表面吸附和迁移的第一性原理研究. 包头: 内蒙古科技大学, 2015.LI P A. First-Principle Studies on Adsorption and Diffusion Behavior of Li on Graphene, BCx and CxN Surfaces. Baotou: Inner Mongulia University of Science & Technology, 2015.

图 1 单层MoSi2N4的3×3×1超胞俯视图(a)和侧视图(b)
Figure 1 Top view (a) and side view (b) of MoSi2N4 monolayer with 3×3×1 supercell

图 2 单层MoSi2N4的3×3×1超胞的硼掺杂位点
Figure 2 Boron-doping sites of MoSi2N4 monolayer with 3×3×1 supercell

图 4 锂离子吸附在构型Ⅰ表面不同位点的三维电子密度差分图
Figure 4 Three-dimensional charge density difference diagram of different Li-ion adsorption sites on the surface of configuration Ⅰ

图 5 构型Ⅰ表面锂离子在不同吸附位点的吸附势能
Figure 5 Adsorption potential energy of different Li-ion adsorption sites on the surface of configuration Ⅰ

图 6 锂离子在构型Ⅰ表面扩散的路径和势垒
Figure 6 Diffusion pathway and diffusion barrier of Li-ion on the surface of configuration Ⅰ
表 1 不同硼掺杂MoSi2N4的3×3×1超胞构型的晶格常数及形成能
Table 1. Lattice constants and formation energy of different configurations of boron-doped MoSi2N4 monolayer with 3×3×1 supercell
| Doped mode | Configuration | Lattice constant | Cell volume / nm3 | Formation energy / eV | ||
| a /nm | b /nm | c /nm | ||||
| Substitution | Ⅰ | 0.876 | 0.876 | 1.640 | 1.258 | -2.02 |
| Ⅱ | 0.870 | 0.870 | 1.940 | 1.468 | -0.40 | |
| Adsorption | Ⅲ | 0.875 | 0.875 | 1.975 | 1.512 | 5.11 |
| Ⅳ | 0.874 | 0.874 | 1.936 | 1.479 | 5.57 | |
| Interstitial | Ⅴ | 0.873 | 0.872 | 1.983 | 1.510 | 4.69 |
| Ⅵ | 0.882 | 0.882 | 1.972 | 1.534 | 5.80 | |
 下载: 导出CSV
下载: 导出CSV
表 2 锂离子在构型Ⅰ表面不同吸附位点的吸附能
Table 2. Adsorption energy of different Li-ion adsorption sites on the surface of configuration Ⅰ
| Li-ion adsorption site | Li-ion adsorption energy (before doped) / eV | Li-ion adsorption energy (after doped) / eV | Distance between Li-ion and boron / nm |
| A | 1.19 | -1.54 | 0.226 |
| B | 1.13 | -1.60 | 0.247 |
| C | 1.12 | -1.66 | 0.292 |
| D | 1.26 | -1.78 | 0.261 |
| E | 1.19 | -1.87 | 0.361 |
| F | 1.12 | -1.72 | 0.407 |
| G | 1.26 | -1.88 | 0.483 |
| H | 1.13 | -1.76 | 0.439 |
| I | 1.19 | -1.91 | 0.548 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: