Table 1.
Crystallographic and structural refinement details for compounds 1 and 2
Citation:
Yu-ting YANG, Zhang-zheng TU, Fan WANG, Yue ZHANG, Jian-ling WANG, Hong-ju YIN, Fei-xiang CHENG. Flexible Bis(imidazole)-Based Ligands Oriented Co(Ⅱ) -Glutarate Metal-Organic Frameworks: Syntheses, Structures and Properties[J]. Chinese Journal of Inorganic Chemistry,
2021, 37(4): 735-743.
doi:
10.11862/CJIC.2021.086

柔性双咪唑配体导向的Co(Ⅱ) -戊二酸金属有机骨架材料的合成、结构和性能
摘要:
在水热条件下合成并表征了2例基于柔性配体构筑的Co(Ⅱ) -MOFs: {[Co(glu)(bimb)]·4H2O}n (1), [Co(glu)(bix)0.5]n (2)(H2glu=戊二酸; bimb=1, 4-双(咪唑-1-基)-丁烷; bix=1, 4-双(咪唑-1-基-亚甲基)-苯)。配合物1显示非穿插的(4, 4)-菱形网格, 通过层间氢键和π…π作用拓展为三维超分子结构。加热失水后1的结构不可逆地降解。配合物2呈现典型的柱-层状三维结构, 其拓扑符号为41263, 中心Co(Ⅱ) 离子采用四方锥构型。此外, 性质研究表明: 配合物1在水溶液中对染料甲基橙具有显著的吸附活性, 配合物2表现出弱的反铁磁行为。
English
Flexible Bis(imidazole)-Based Ligands Oriented Co(Ⅱ) -Glutarate Metal-Organic Frameworks: Syntheses, Structures and Properties
Abstract:
Two new Co(Ⅱ) -MOFs based on flexible ligands, namely {[Co(glu)(bimb)]·4H2O}n(1) and [Co(glu)(bix)0.5]n(2)(H2glu=glutaric acid; bimb=1, 4-bis(imidazol-1-yl)-butane; bix=1, 4-bis(imidazol-1-yl-methylene)-benzene), have been hydrothermally synthesized and structurally characterized. Compound 1 displays non-interpenetrated(4, 4)-rhomboid grid layers. A 3D supramolecular framework is invoked through interlayer hydrogen bonding and π…π interactions. Upon dehydration the structure of 1 degraded irreversibly. Compound 2 exhibits a typical pillar-layered 3D framework with 41263 topology, in which the Co(Ⅱ) ion adopts a square pyramidal coordination geometry. Moreover, Compound 1 exhibited significant adsorption activity towards methyl orange in aqueous solution, and 2 showed weak antiferromagnetic behavior. CCDC: 2012753, 1; 2012754, 2.
-
Key words:
- crystal structure
- / glutaric acid
- / bis(imidazole) ligands
- / adsorption
- / magnetism
-
0. Introduction
Metal-organic frameworks (MOFs) with various intriguing structural motifs have been extensively studied for their versatile chemical and physical properties, as well as potential applications in luminescence, catalysis, magnetism, gas storage and separation, solvent exchange, and so on[1-6].Generally, the construction of functional MOFs with attractive topological structures is primarily dependent upon judicious selection of well-designed organic ligands.According to this consideration, vast numbers of organic ligands have been widely employed.So far, plenty of research efforts have been devoted to MOFs assembled by rigid aromatic polycarboxylate ligands owing to their rich coordination modes and stable configuration[7-8].However, compared with aromatic carboxylate ligands, little attention has been paid to aliphatic carboxylate ligands even though their flexibility and conformational freedoms can offer a greater degree of structural diversity[9-12].Therefore, much more investigations of the correlation between the conformation of flexible ligands and the final topologies of the MOFs need to be done and a rational design would be a long-range challenge.
In our previous report, we investigated a series of MOFs constructed by different aromatic carboxylate ligands with the bis(imidazole) ligands[13-15].As a continuation of this attractive subject, we turned our attention to study the coordination behavior of aliphatic dicarboxylates in combination with the flexible 1, 4-bis(imidazol-1-yl)-butane (bimb) and 1, 4-bis(imidazol-1-yl-methene-yl)-benzene (bix).The—(CH2)4—alkyl spacer in the bimb ligand permits two imidazole groups to twist freely to meet the geometry requirements of metal centers.This flexibility induces different conformations.And the bix ligand possesses cis and trans conformations as a result of the free rotation of two methylene groups.Both of them have been justified as good candi-dates for structural assembly of unpredictable and interesting MOFs[16-18].Therefore, we investigated two interesting Co(Ⅱ) -MOFs, namely{[Co(glu)(bimb)]·4H2O}n (1) and[Co(glu)(bix)0.5]n(2), constructed by two flexible bis(imidazole) ligands with different conformations as auxiliary ligands in the presence of glutaric acid (H2glu) as coligands.The two compounds display substantially different structural morphologies depending on the binding modes of the glutarate dianion, the coordination environment of the Co(Ⅱ) ions, and the length and conformations of the bis(imidazole) ligands.In addition, compound 1 displayed unique selective adsorption to MO (methyl orange) dyes, and 2 had a weak antiferromagnetic performance.
1. Experimental
1.1 Materials and measurements
All commercially available chemicals were of reagent grade and were used as received without further purification.The ligands bimb and bix were synthesized by literature procedures[19].The infrared spectra were performed on a Varian FT-IR 640 spectrometer with KBr pellets in the 400~4 000 cm-1 region.Elemental analyses were measured on a Perkin-Elmer2400 C H N elemental analyzer (C, H and N).Powder X-ray diffraction (PXRD) patterns were collected on a Rigaku D/MAX-ⅢC powder diffractometer at 40 kV, 40 mA with Cu Kα radiation (λ=0.154 06 nm) and a graphite monochromator scanning from 5° to 30°.The thermogravimetric analysis (TGA) was performed with a Shimadzu TGA-50H TG analyzer in a range of 25~800 ℃ under a nitrogen flow at a heating rate of 5 ℃ ·min-1.Magnetic susceptibility measurements were carried out on a Quantum Design MPMSXL SQUID magnetometer and PPMS-9T system.A correction was made for the diamagnetic contribution prior to data analysis.UV-Vis spectra studies were performed at room temperature on a Perkin Elmer Lambda 900 UV/Vis/NIR spectrophotometer equipped with an integrating sphere in a wavelength range of 200~1 200 nm.
1.2 Synthesis
1.2.1 Synthesis of{[Co(glu)(bimb)]·4H2O}n(1)
The mixture of Co Cl2·6H2O (0.24 g, 1.0 mmol), glutaric acid (0.13 g, 1.0 mmol) and bimb (0.19 g, 1.0 mmol) was dissolved in 8 mL of distilled water.The pH value was then adjusted to 6.0 with 1 mol·L-1 NaOH solution and the resulting mixture was transferred and sealed in a 25 mL Teflon-lined stainless-steel vessel, heated at 160 ℃ for 72 h, and then cooled to room temperature over 24 h.Red block-shaped crystals were filtered off and dried in air.Yield: 63%(based on Co).Elemental analysis Calcd.for C15H28Co N4O8(%): C 39.92, H 6.25, N 12.41.Found(%): C 39.25, H 5.89, N 13.36.IR (cm-1): 3 142(m), 2 964(m), 1 551(s), 1 405(s), 1 291(m), 1 229(m), 1 079(s), 945(m), 835(m), 723(m), 653(m).
1.2.2 Synthesis of[Co(glu)(bix)0.5]n (2)
The reaction conditions of 2 were similar to that described for 1 except that bix was used instead of bimb.Red block crystals were obtained in 65%yield based on Co(Ⅱ) Elemental analysis Calcd.for C12H13Co N2O4(%): C 46.77, H 4.25, N 9.09;Found: C 47.22, H 4.13, N 8.45.Selected IR (KBr): 3 168(m), 3 150(m), 2 946(m), 1 601(s), 1 451(s), 1 405(s), 1 309(m), 1 229(m), 1 095(s), 955(m), 839(m), 723(m), 661(m).
1.3 X-ray crystallography and structure determination
Single crystals suitable for X-ray analyses were used on a Bruker SMART APEX vⅡ CCD diffractome-ter using graphite monochromated Mo Kα radiation (λ=0.071 073 nm) by using a φ-ω scan technique at room temperature.Lorentz polarization and absorption corrections were applied.The structures were solved by direct methods and refined with the full-matrix least-squares technique using the SHELXS-97 and SHELXL-97 programs[20-21].The hydrogen atoms were assigned with common isotropic displacement factors and included in the final refinement by use of geometrical restrains, while the non-hydrogen atoms were treated with common anisotropic displacement factors and included in the final refinement with geometrical restrains.The crystallographic data and details of the structure determinations for compounds 1 and 2 are listed in Table 1.Selected bond lengths and angles for 1 and 2 are given in Table 2 and 3.
Table 1

 下载:
导出CSV
下载:
导出CSV
Compound 1 2 Formula C15H28CoN4O8 C12H13CoN2O4 Formula weight 451.34 308.17 T/K 293(2) 293(2) Crystal system Monoclinic Monoclinic Space group C2/c P21/n a / nm 0.931 70(19) 1.014 5(2) b / nm 1.445 7(3) 0.802 96(16) c / nm 1.574 9(3) 1.505 2(3) β /(°) 101.77(3) 106.33(3) V /nm3 2.076 7(7) 1.176 7(4) Z 4 4 Dc/(g·cm-3) 1.444 1.740 F(000) 948 632 Absorption coefficient / mm-1 0.874 1.470 θ for data collection 3.11~27.48 2.17~27.94 Reflection collected, unique 9 959, 2 356 11 628, 2 809 GOF on F2 1.002 1.002 R1, wR2 [I>2σ(I)]* 0.043 5, 0.116 4 0.027 4, 0.072 2 Largest diff. peak and hole/ (e·nm-3) 467 and -427 415 and -366 $* R_{1}=\sum\left\|F_{0}|-| F_{\mathrm{c}}\right\| /\left|F_{\mathrm{o}}\right|, w R_{2}=\left[\sum w\left(F_{\mathrm{o}}^{2}-F_{\mathrm{c}}^{2}\right)^{2} / \sum w\left(F_{\mathrm{o}}^{2}\right)^{2}\right]^{1 / 2}$ Table 2
Table 2. Selected bond lengths (nm) and bond angles (°) for 1下载:
导出CSV
Co1—N1 0.205 4(2) Co1—N1#1 0.205 4(2) Co1—O2 0.212 77(18) Co1—O2#1 0.212 77(18) Co1—O1#1 0.228 9(2) Co1—O1 0.228 9(2) N1#1—Co1—O2#1 97.73(8) N1#1—Co1—02 98.30(8) N1—Co1—02 97.73(8) O2#1—Co1—O2 154.89(12) N1#1—Co1—01#1 92.66(8) N1—Co1—01#1 154.85(8) O2#1-Co1-O1#1 58.30(7) O2-Co1-O1#1 101.71(8) N1-Co1-01 92.66(8) N1#1—Co1—O1 154.85(8) O2#1—Co1—O1 101.71(8) O2—Co—O1 58.30(7) O1#1—Co1—O1 84.25(12) N1—Co1—N1#1 100.19(11) N1—Co1—O2#1 98.30(8) Symmetry codes: #1: -x, y, -z+3/2; #2: -x+1, -y+1, -z+2; #3: -x+1, y, -z+3/2 for 1 Table 3
Table 3. Selected bond lengths (nm) and bond angles (°) for 2下载:
导出CSV
Co1—O4 0.201 40(12) Co1—O1 0.203 24(14) Co1—O3 0.203 35(12) Co1—N1 0.204 35(15) Co1—O2 0.213 77(14) Co1—Co1#1 0.283 38(7) O4—Co1—O1 89.97(5) O4—Co1—O3 162.68(5) O1—Co1—O3 90.01(5) O4—Co1—N1 103.45(5) O1—Co1—N1 105.75(6) O3—Co1—N1 93.22(6) O4—Co1—O2 88.32(5) O3—Co1—O2 86.84(5) O1—Co1—O2 163.66(5) N1—Co1—O2 90.45(6) O4—Co1—Co1#1 82.79(4) O1—Co1—Co1#1 95.80(4) O3—Co1—Co1#1 79.97(4) N1—Co1—Co1#1 157.45(4) O2—Co1—Co1#1 67.86(4) Symmetry codes: #1: -x+1, -y+1, -z; #2: x+1/2, -y+1/2, z+1/2; #3: -x+1, -y+1, -z+1; #4: x-1/2, -y+1/2, z-1/2 for 2. CCDC: 2012753, 1; 2012754, 2.
2. Results and discussion
2.1 Structural description of 1
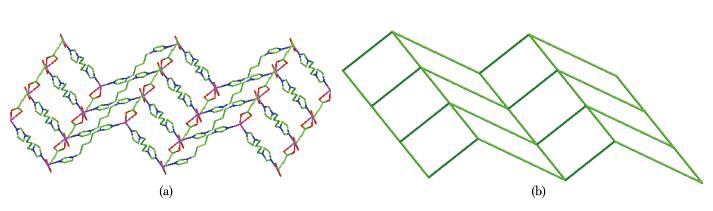
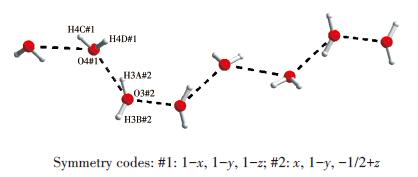
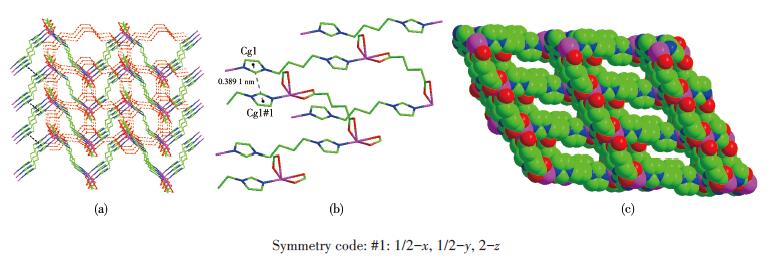
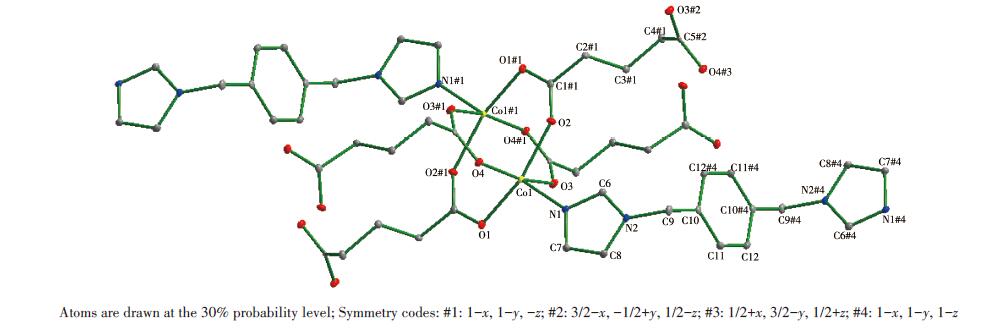
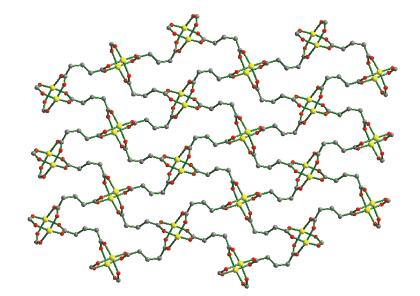
Single-crystal X-ray diffraction analysis reveals that compound 1 belongs to the monoclinic space group of C2/c.The fundamental building unit of 1 contains one independent Co(Ⅱ) cation, one glu2- anion, one bimb ligand, and four lattice water molecules(Fig. 1).Each Co(Ⅱ) ion is six-coordinated and exhibits a distorted octahedral geometry[Cd N2O4], which is surrounded by four carboxylic oxygen atoms from two different glu2- anions and two nitrogen atoms from two bimb ligands.In compound 1, the glu2- ligand displays a μ2-bridging mode, in which both of the carboxylate groups are in the μ1-η1∶η1 chelating-bridging mode.The neighboring Co(Ⅱ) ions are conjoined both through bis-chelating glu2- anion and bimb ligand to construct a normal (4, 4) rhomboid grid layer (Fig. 2a, 2b), propagating parallel to the ac crystal plane.The Co…Co distances around the grid perimeters are 0.931 7 and 1.415 6 nm, through the shorter glu2- and longer bimb ligands, respectively.The lattice water molecules (O3, O4) interpenetrate between layers resulting in wavy 1D infinite chains by hydrogen bonds (Fig. 3).As shown in Fig. 4a, there are also significant hydrogen-bonding interactions between water molecules and the oxygen atoms (O1, O2) of the carboxyl groups from glu2- ligands (O3…O1, 0.286 4 nm, O3—H3B…O1, 150.40°; O4…O2, 0.278 1 nm, O4—H4D…O2, 138.67°, Table 4).Moreover, the imidazole rings of bimb ligandsbetween the neighboring layers are obviously parallel with each other, and the imidazole…imidazole rings stack in a parallel-displaced form (π…π stacking offset distance is 0.389 1 nm)(Fig. 4b), implying weak intermolecular π…π stacking interactions.If these interactions are treated as “bonds”, a 3D supramolecular network can be invoked.Moreover, 1D irregular rhomboid shaped microporous channels exist along the b direction (Fig. 4c), and the ratio of accessible pore volume in one unit cell is 11.2%, calculated by PLA-TON[22].The accessible volume was occupied by water molecules.
Figure 1
Figure 2
 Figure 2. (a) Single[Co(glu)(bimb)]nlayer in 1; (b) Schematic representation of (4, 4) rhomboid grid layer
Figure 2. (a) Single[Co(glu)(bimb)]nlayer in 1; (b) Schematic representation of (4, 4) rhomboid grid layerFigure 3
Figure 4
 Figure 4. (a) 3D supramolecular network expanded by hydrogen bonds (red dotted) and π…π stacking interactions(black dotted, only representative were displayed for clarity); (b)π…π interactions between imidazole rings of bimb; (c) View of 1D rhombus microporous channels
Figure 4. (a) 3D supramolecular network expanded by hydrogen bonds (red dotted) and π…π stacking interactions(black dotted, only representative were displayed for clarity); (b)π…π interactions between imidazole rings of bimb; (c) View of 1D rhombus microporous channelsTable 4
Table 4. H⁃bonding parameters for 1下载:
导出CSV
D—H—A d(D—H) / nm d(H — A)/nm d(D—A) / nm ∠DHA / (°) O3—H3A—O1#1 0.089 98 0.235 2 0.286 5 159.49 O3—H3B—O4#1 0.084 67 0.207 5 0.262 8 118.63 O4—H4C—O3#2 0.086 56 0.1770 0.262 8 170.63 O4—H4D—O2#2 0.095 26 0.199 3 0.278 1 138.68 Symmetry codes: #1: 1-x, 1-y, 1-z; #2: x, 1-y, -1/2+z. 2.2 Structural description of 2
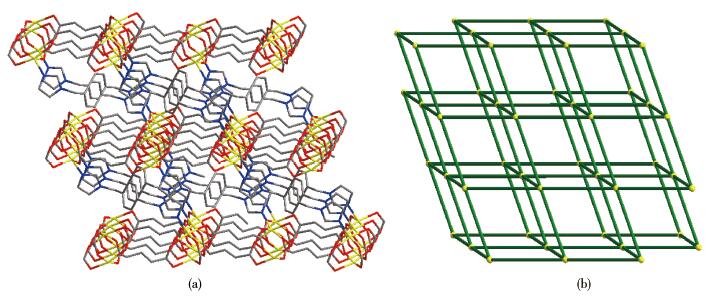
Single-crystal X-ray diffraction analysis reveals that compound 2 belongs to the space group P21/n.The fundamental building unit of 2 contains one independent Co(Ⅱ) cation, one glu2- anion, and half a bix ligand(Fig. 5).Each Co(Ⅱ) ion is five-coordinated and exhibits a distorted square pyramid geometry [CoNO4], which is surrounded by four carboxylic oxygen atoms from four different glu2- ligands and one imidazolyl nitrogen donor from the bix ligand.The glu2- ligand acts as a μ4-bridging linker with the two carboxylate groups in the same μ2-η1∶η1 monodentate fashion, which is different from compound 2.The adjacent Co1 and Co#1 were bridged by four carboxyl groups from different glu2- ligands, resulting in paddlewheel dinuclear[Co2(COO)4]second building unit (SBU).The distance of Co…Co is 0.283 4 nm.On the basis of this connection mode, the glu2- ligands connect adjacent Co(Ⅱ) ions resulting in a neutral 2D[Co(glu)]n layer running parallel to the ab plane (Fig. 6).Furthermore, these 2D layers are pillared by trans-bix ligands into a 3D pillared-layered structure (Fig. 7a).Each [Co2(COO)4] SBU is linked to six adjacent ones through four glu2- anions and two bix ligands.As a result, a 6-connected pcu net with a Schläfli symbol of 41263 is formed.The schematic description of this network is shown in Fig. 7b.
Figure 5
Figure 6
Figure 7
 Figure 7. (a) Perspective view of pillared-layered 3D structure of 2; (b) Schematic representation of pcu framework of 2
Figure 7. (a) Perspective view of pillared-layered 3D structure of 2; (b) Schematic representation of pcu framework of 22.3 Thermogravimetric analysis and PXRD
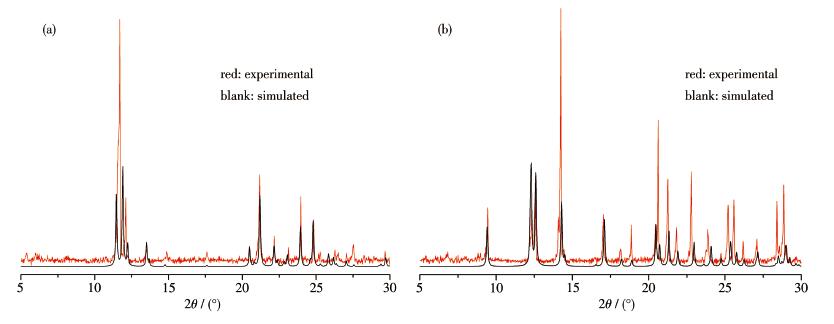
In order to check the phase purity of these compounds, the PXRD patterns of compounds 1 and 2 were checked at room temperature.As shown in Fig. 8, the peak positions of the simulated and experimental PXRD patterns are in agreement with each other, demonstrating the good phase purity of the compounds.The differences in intensity may be due to the preferred orientation of the crystalline powder samples.
Figure 8
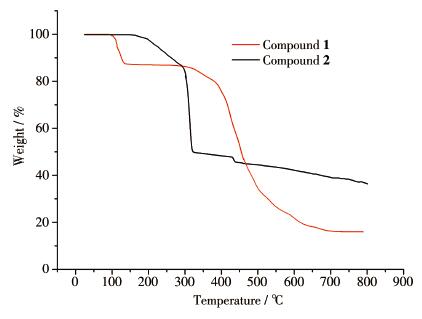
To characterize the compounds more fully in terms of thermal stability, their thermal behaviors were studied by TGA (Fig. 9).The experiments were performed on crystalline samples from 25 to 800 ℃ under a nitrogen atmosphere at a heating rate of 5 ℃ ·min-1.For 1, The TGA curve showed that the first weight loss occurred between 99 and 138 ℃, which corresponds to the loss of lattice water molecules (Calcd.15.9%, Obsd.13.8%).As shown in Fig. 9, the framework of 1 without lattice water molecule can maintain stability in a range of 138~310 ℃.Then further weight loss was observed at the temperature of 310 ℃.Compound 2 can be stable up to 163 ℃.Upon further heating, the structure begins to collapse, following the release of the ligand molecules.
Figure 9
2.4 Adsorption of dyes
Recently, it is very popular to adsorb and separate dye molecules from liquid phase by employing porous MOFs.The main factors affecting the dye adsorption performance are electrostatic attraction, size selection, and ion exchange[23].Because compound 1 is electrically neutral, we speculate that the adsorption procedure may be largely governed by their size.The different size and electrical dyes, such as MO, bromophenol blue (BB), methylene blue (MB), cresol red (CR), and rhodamine B (RB) were selected to study the sorption of 1.
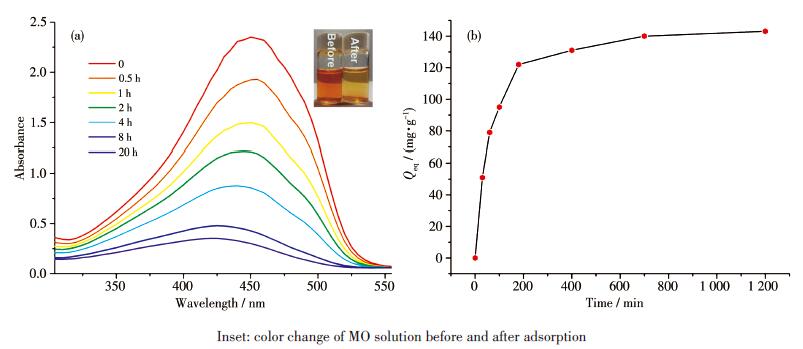
In view of the previous experimental exploration and sample preparation[24-25], 30 mg of 1 was dispersed in the aqueous solution of various dyes with the concentration of 30 mg·L-1 in flat-botton glass flasks, and then the mixture was shaken at a constant shaking speed of 200 r·min-1.The selective adsorption behaviors were investigated by UV-Vis spectrophotometry and photographs.As can be seen from Fig. 10, the color of compound 1 immersed in MO appears to fade obviously after 120 min, whereas the color of other solutions did not change significantly.It was concluded that1has a strong capability and high efficiency for adsorption of MO.The size of 1 is 0.931 7 nm×1.415 6nm, whereas the molecular size of the methyl orange dye is about 1.54 nm×0.46 nm×0.47 nm.The dye molecules can be captured when the size of dyes well matches the porosities of the sophisticated framework in any two directions.
Figure 10
 Figure 10. Color differences of various dyes before and after addition of 1 in aqueous solution
Figure 10. Color differences of various dyes before and after addition of 1 in aqueous solutionThe quantity of adsorbed dye could be calculated at equilibrium by the formula[26]: Qeq=(c0-ceq)V/m, where c0 and ceq represent the initial and the equilibrium concentrations of the dye (mg·L-1), respectively, and m and V represent the mass of adsorbent (g) and solution volume (L), respectively.By calculation, the value of Qeq for MO was 143 mg·g-1 for 1(Fig. 11).
Figure 11
 Figure 11. (a) UV-Vis spectra of MO exhibiting the adsorption processes with 1 at different times; (b) Time-resolved adsorption capacity of MO on compound 1
Figure 11. (a) UV-Vis spectra of MO exhibiting the adsorption processes with 1 at different times; (b) Time-resolved adsorption capacity of MO on compound 12.5 Magnetic properties
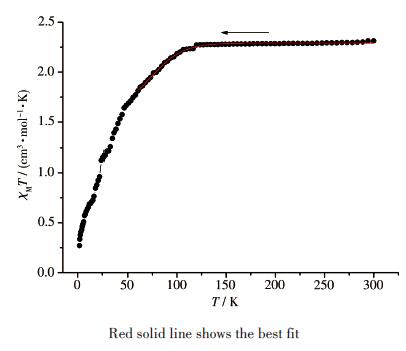
The temperature-dependent magnetic susceptibility data of compound 2 was measured for polycrystalline samples at an applied magnetic field of 1 000 Oe in a temperature range of 2~300 K.As shown in Fig. 12, as the temperature was lowered, the χMT value exhibited a monotonic decrease down to 0.54 cm3·K·mol-1 at 2.0 K, indicating that antiferromagnetic interactions are operative and the ground state is nonmagnetic in 2.
Figure 12
The χMT value of 2 at 300 K was 2.31 cm3·K·mol-1, which was obviously larger than one isolated spinonly Co(Ⅱ) ion (1.875 cm3·K·mol-1).This larger value is the result of contributions to the susceptibility from orbital angular momentum at high temperatures[27].The Co…Co separation across adjacent Co(Ⅱ) ions bridged by the four carboxylic groups was 0.283 4 nm, significantly shorter than 1.331 8 and 8.335 nm for the bix and the shortest two carboxylic group bridges.Thus, the main magnetic interactions may be considered to occur between adjacent Co(Ⅱ) ions, whereas the superexchange interactions between Co(Ⅱ) ions through glu2- and bix bridges can be ignored due to the long Co…Co separations.
We tried to analyze the magnetic data using the isotropic one-ion approximation with the spin-orbit coupling in Oh symmetry, and Ĥ=-(3/2)κλLS+β[-(3/2)κL+geS]H.A magnetic susceptibility equation was obtained as follows[30-31]:
$\chi_{\mathrm{M}}=\frac{N \beta^{2}}{k_{\mathrm{B}} T} \frac{F_{1}}{F_{2}}$ $F_{1}=\frac{63}{20}(-2+\kappa)^{2} x+\frac{2(4+3 \kappa)^{2}}{25 \kappa}+ \\ \left[\frac{2}{45}(11-3 \kappa)^{2} x+\frac{88(4+3 \kappa)^{2}}{2205 \kappa}\right] \exp \left(-\frac{15 \kappa}{4} x\right)+ \\ \left[\frac{1}{36}(10+3 \kappa)^{2} x-\frac{10(4+3 \kappa)^{2}}{81 \kappa}\right] \exp (-6 \kappa x)$ $F_{2}=x\left[3+2 \exp \left(-\frac{15 \kappa}{4} x\right)+\exp (-6 \kappa x)\right]$ where x=λ/(kT), λis the spin-orbit coupling; κ is the orbital reduction factor, and other symbols have their usual meanings.The data were well simulated in a temperature range of 60~300 K for compound 2, and calculated curves are shown in Fig. 12.Fitting parameters were κ=0.91, λ=-105 cm -1.The g values were calculated as 2.20.Agreement factors defined as R=∑[(χMT)exp-(χMT)calc]2/∑[(χMT)exp]2 were 2.31×10-4.However, thecryomagnetic data could not be explained by theseequations.
3. Conclusions
Two Co-MOFs have been obtained by different flexible bis(imidazole) ligands and glutaric acid under the same hydrothermal condition.The comparison of compounds 1 and 2 demonstrates that the structure diversities may be greatly induced by coordination modes of the glutarate dianions, the coordination environment about the center Co(Ⅱ) ions, and the length and the conformation of the bis(imidazole) ligands.Compound 1 exhibits much better selective adsorption properties to MO than the adsorption of other dyes in an aqueous solution.The excellent selective adsorption properties, as well as its properties of ease of preparation and convenient manipulation, make it highly valuable for potential applications in dyerelated environmental pollution.The magnetic susceptibility measurements indicate that compound 2 exhibits antiferromagnetic coupling between adjacent Co(Ⅱ) ions in [Co2(COO)4] unit.Further systematic works for the design and synthesis of such crystalline materials with long-chain aliphatic acid, N-donor auxiliary ligands, and other metal ions, also the related properties investigations are now in progress to explore the desirable materials.
-
-
[1]
Cadiau A, Kolobov N, Srinivasan S, Goesten M G, Haspel H, Bavykina A V, Tchalala M R, Maity P, Goryachev A, Poryvaev A S, Eddaoudi M, Fedin M V, Mohammed O F, Gascon J. Angew. Chem. Int. Ed. , 2020, 59: 1-6 doi: 10.1002/anie.201914874
-
[2]
Tian X M, Yao S L, Qiu C Q, Zheng T F, Chen Y Q, Huang H, Chen J L, Liu S J, Wen H R. Inorg. Chem. , 2020, 59: 2803-2810 doi: 10.1021/acs.inorgchem.9b03152
-
[3]
田千, 张建国, 倪春燕, 杨战永, 姚志刚, 张敏杰, 郎建平. 无机化学学报, 2020, 36(6): 1063-1071 https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htmTIAN Q, ZHANG J G, NI C Y, YANG Z Y, YAO Z G, ZHANG M J, LANG J P. Chinese J. Inorg. Chem. , 2020, 36(6): 1063-1071 https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htm
-
[4]
Yao S L, Zheng T F, Tian X M, Liu S J, Cao C, Zhu Z H, Chen Y Q, Chen J L, Wen H R. CrystEngComm, 2018, 20: 5822-5832 doi: 10.1039/C8CE01261J
-
[5]
Wu H Q, Yan C S, Luo F, Krishna R. Inorg. Chem. , 2018, 57: 3679-3682 doi: 10.1021/acs.inorgchem.8b00341
-
[6]
Li J R, Ma Y G, McCarthy C M, Sculley J, Yu J, Jeong H K, Balbuena P B, Zhou H C. Coord. Chem. Rev. , 2011, 255: 1791-1823
-
[7]
Cook T R, Zheng Y R, Stang P J. Chem. Rev. , 2013, 113: 734-777
-
[8]
Lu W G, Wei Z W, Gu Z Y, Liu T F, Park J, Park J, Tian J, Zhang M W, Zhang Q, Gentle T, Bosch M, Zhou H C. Chem. Soc. Rev. , 2014, 43: 5561-5593
-
[9]
Yang J X, Qin Y Y, Ye R P, Zhang X, Yao Y G. CrystEngComm, 2016, 18: 8309-8320 doi: 10.1039/C6CE01607C
-
[10]
Martin D P, Montney M R, Supkowski R M, LaDuca R L. Cryst. Growth Des. , 2008, 8: 3091-3097 doi: 10.1021/cg8003118
-
[11]
Wang Z, Xing Y H, Wang C G, Sun L X, Zhang J, Ge M F, Niu S Y. CrystEngComm, 2010, 12: 762-773 doi: 10.1039/B916127A
-
[12]
Gao D Z, Wei H X, Wang X G, Liu Z Y, Sun Y Q, Zhang G Y, Xu Y Y. Inorg. Chem. Commun. , 2013, 38: 54-57 doi: 10.1016/j.inoche.2013.10.020
-
[13]
Yang Y T, Tu C Z, Yin H J, He C X, Zhao Q, Cheng F X. Eur. J. Inorg. Chem. , 2019, 2019: 4582-4591 doi: 10.1002/ejic.201900930
-
[14]
Yang Y T, Tu C Z, Xu X L, Xu L L, Yan B L, Cheng F X. Aust. J. Chem. , 2019, 72: 341-346 doi: 10.1071/CH18459
-
[15]
Yang Y T, Tu C Z, Miao J J, Li J L, Chen G, Yin H J, Cheng F X. Z. Anorg. Allg. Chem. , 2015, 641: 2576-2580 doi: 10.1002/zaac.201500232
-
[16]
Yang J X, Qin Y Y, Cheng J K, Yao Y G. Cryst. Growth Des. , 2015, 15: 2223-2234
-
[17]
李秀梅, 杨佳琦, 潘亚茹, 刘博, 周实. 无机化学学报, 2020, 36(4): 730-736 https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htmLI X M, YANG J Q, PAN Y R, LIU B, ZHOU S. Chinese J. Inorg. Chem. , 2020, 36(4): 730-736 https://www.cnki.com.cn/Article/CJFDTOTAL-DLXB201502001.htm
-
[18]
Liu G X, Zhou H, Wang X F. J. Chem. Crystallogr. , 2013, 43: 335-341
-
[19]
Hoskins B F, Robson R, Slizys D A. J. Am. Chem. Soc. , 1997, 119: 2952-2953
-
[20]
Sheldrick G M. SHELXS-97, Program for Crystal Structure Solution, University of Göttingen, Germany, 1997.
-
[21]
Sheldrick G M. SHELXL-97, Program for the Refinement of Crystal Structures from Diffraction Data. University of Göttingen, Germany, 1997.
-
[22]
Spek A L. PLATON, a Multipurpose Crystallographic Tool, Utrecht University, The Netherlands, 2002.
-
[23]
Wu D, Liu J, Jin J, Cheng J G, Wang M, Yang G P, Wang Y Y. Cryst. Growth Des. , 2019, 19: 6774-6783
-
[24]
Wan S H, Li L, Liu J M, Liu B, Li G H, Zhang L R, Liu Y L. Cryst. Growth Des. , 2020, 20: 3199-3207
-
[25]
Jin J, Yang G P, Liu Y C, Cheng S, Liu J, Wu D, Wang Y Y. Inorg. Chem. , 2019, 58: 339-348
-
[26]
Yang L, Li X, Sun C Y, Wu H, Wang C G, Su Z M. New J. Chem. , 2017, 41: 3661-3666
-
[27]
Ma L F, Wang L Y, Wang Y Y, Batten S R, Wang J G. Inorg. Chem. , 2009, 48: 915-924
-
[28]
Liu Y H, Tsai H L, Lu Y L, Wen Y S, Wang J C, Lu K L. Inorg. Chem. , 2001, 40: 6426-6431
-
[29]
Chen P K, Qi Y, Che Y X, Zheng J M. CrysEngComm, 2010, 12: 720-724
-
[30]
Figgis B N, Gerloch M, Lewis J, Mabbs F E, Webb G A. J. Chem. Soc. A, 1968: 2086-2093
-
[31]
Sakiyama H, Ito R, Kumagai H, Inoue K, Sakamoto M, Nishida Y, Yamasaki M. Eur. J. Inorg. Chem. , 2001: 2027-2032
-
[1]
-
Figure 2 (a) Single[Co(glu)(bimb)]nlayer in 1; (b) Schematic representation of (4, 4) rhomboid grid layer
Figure 4 (a) 3D supramolecular network expanded by hydrogen bonds (red dotted) and π…π stacking interactions(black dotted, only representative were displayed for clarity); (b)π…π interactions between imidazole rings of bimb; (c) View of 1D rhombus microporous channels
Figure 7 (a) Perspective view of pillared-layered 3D structure of 2; (b) Schematic representation of pcu framework of 2
Figure 10 Color differences of various dyes before and after addition of 1 in aqueous solution
Figure 11 (a) UV-Vis spectra of MO exhibiting the adsorption processes with 1 at different times; (b) Time-resolved adsorption capacity of MO on compound 1
Table 1. Crystallographic and structural refinement details for compounds 1 and 2
Compound 1 2 Formula C15H28CoN4O8 C12H13CoN2O4 Formula weight 451.34 308.17 T/K 293(2) 293(2) Crystal system Monoclinic Monoclinic Space group C2/c P21/n a / nm 0.931 70(19) 1.014 5(2) b / nm 1.445 7(3) 0.802 96(16) c / nm 1.574 9(3) 1.505 2(3) β /(°) 101.77(3) 106.33(3) V /nm3 2.076 7(7) 1.176 7(4) Z 4 4 Dc/(g·cm-3) 1.444 1.740 F(000) 948 632 Absorption coefficient / mm-1 0.874 1.470 θ for data collection 3.11~27.48 2.17~27.94 Reflection collected, unique 9 959, 2 356 11 628, 2 809 GOF on F2 1.002 1.002 R1, wR2 [I>2σ(I)]* 0.043 5, 0.116 4 0.027 4, 0.072 2 Largest diff. peak and hole/ (e·nm-3) 467 and -427 415 and -366 $* R_{1}=\sum\left\|F_{0}|-| F_{\mathrm{c}}\right\| /\left|F_{\mathrm{o}}\right|, w R_{2}=\left[\sum w\left(F_{\mathrm{o}}^{2}-F_{\mathrm{c}}^{2}\right)^{2} / \sum w\left(F_{\mathrm{o}}^{2}\right)^{2}\right]^{1 / 2}$  下载: 导出CSV
下载: 导出CSV
Table 2. Selected bond lengths (nm) and bond angles (°) for 1
Co1—N1 0.205 4(2) Co1—N1#1 0.205 4(2) Co1—O2 0.212 77(18) Co1—O2#1 0.212 77(18) Co1—O1#1 0.228 9(2) Co1—O1 0.228 9(2) N1#1—Co1—O2#1 97.73(8) N1#1—Co1—02 98.30(8) N1—Co1—02 97.73(8) O2#1—Co1—O2 154.89(12) N1#1—Co1—01#1 92.66(8) N1—Co1—01#1 154.85(8) O2#1-Co1-O1#1 58.30(7) O2-Co1-O1#1 101.71(8) N1-Co1-01 92.66(8) N1#1—Co1—O1 154.85(8) O2#1—Co1—O1 101.71(8) O2—Co—O1 58.30(7) O1#1—Co1—O1 84.25(12) N1—Co1—N1#1 100.19(11) N1—Co1—O2#1 98.30(8) Symmetry codes: #1: -x, y, -z+3/2; #2: -x+1, -y+1, -z+2; #3: -x+1, y, -z+3/2 for 1
下载: 导出CSV
Table 3. Selected bond lengths (nm) and bond angles (°) for 2
Co1—O4 0.201 40(12) Co1—O1 0.203 24(14) Co1—O3 0.203 35(12) Co1—N1 0.204 35(15) Co1—O2 0.213 77(14) Co1—Co1#1 0.283 38(7) O4—Co1—O1 89.97(5) O4—Co1—O3 162.68(5) O1—Co1—O3 90.01(5) O4—Co1—N1 103.45(5) O1—Co1—N1 105.75(6) O3—Co1—N1 93.22(6) O4—Co1—O2 88.32(5) O3—Co1—O2 86.84(5) O1—Co1—O2 163.66(5) N1—Co1—O2 90.45(6) O4—Co1—Co1#1 82.79(4) O1—Co1—Co1#1 95.80(4) O3—Co1—Co1#1 79.97(4) N1—Co1—Co1#1 157.45(4) O2—Co1—Co1#1 67.86(4) Symmetry codes: #1: -x+1, -y+1, -z; #2: x+1/2, -y+1/2, z+1/2; #3: -x+1, -y+1, -z+1; #4: x-1/2, -y+1/2, z-1/2 for 2.
下载: 导出CSV
Table 4. H⁃bonding parameters for 1
D—H—A d(D—H) / nm d(H — A)/nm d(D—A) / nm ∠DHA / (°) O3—H3A—O1#1 0.089 98 0.235 2 0.286 5 159.49 O3—H3B—O4#1 0.084 67 0.207 5 0.262 8 118.63 O4—H4C—O3#2 0.086 56 0.1770 0.262 8 170.63 O4—H4D—O2#2 0.095 26 0.199 3 0.278 1 138.68 Symmetry codes: #1: 1-x, 1-y, 1-z; #2: x, 1-y, -1/2+z.
下载: 导出CSV
-















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 1151
- HTML全文浏览量: 131
 下载:
下载:
