Received Date:
10 September 2020 Revised Date:
05 January 2021 Available Online:
10 March 2021
Abstract:
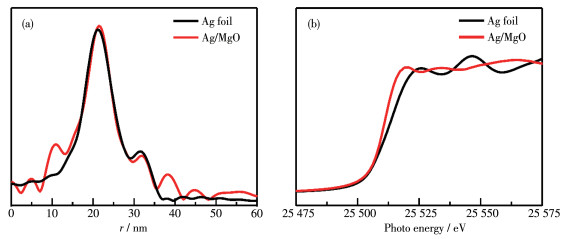
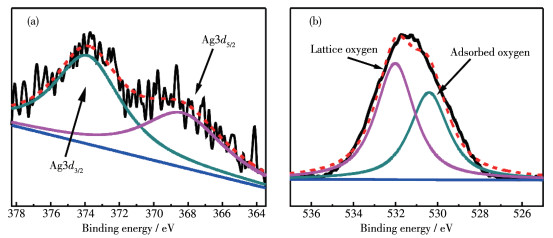
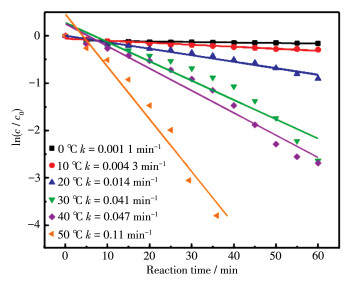
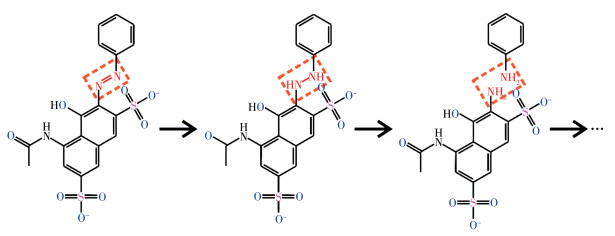
Transition metal supported catalysts were prepared by impregnation method with nano MgO as support. Among several supported transition metals, Ag/MgO showed the best performance in degradation of azo dyes and the structure and micro morphology of selected Ag/MgO catalysts were characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM), extended X-ray absorption fine structure spectroscopy (EXAFS) and X-ray absorption near edge structure (XANES). The results show that Ag was evenly dispersed on the surface of MgO in the form of nano clusters, and the formation of bimetallic sites between Ag and Mg and the high electron density of Ag in the catalysts give it high catalytic activity. In formaldehyde solution, azo dye AR1 could be efficiently degraded at room temperature without illuminating or heating treatment. Temperature and formaldehyde concentration are the main factors affecting the degradation efficiency. The degradation efficiency increased with temperature and the optimal formaldehyde concentration was 1 mol·L-1. Two kinds of free radicals were detected by electron paramagnetic resonance (EPR) trapping experiment to play significant roles in azo dyes degradation process. The reductive hydrogen radical and oxidizing superoxide radical synergistic effect could easily break the chromogenic group (-N=N-) in dye molecules. This "reduction-oxidation" synergistic mechanism improves the reaction efficiency. Moreover, as one of the common pollutants, using aldehyde promoter achieves the "dual treatment of pollutants".
Figure 4.
UV-Vis absorption spectra of AR1 dyes degraded at room temperature by catalysts with different loaded metals:
(a) Ag/MgO, (b) Au/MgO, (c) Pd/MgO, (d) Pt/MgO; (e) Pictures of degraded AR1 solution corresponding to (a)
Ceretta M B, Vieira Y, Wolski E A, Foletto E L, Silvestri S. J. Water Process Eng. , 2020, 35: 101230 doi: 10.1016/j.jwpe.2020.101230
[2]
El-Wakiel N, El-Ghamry H. Int. J. Chem. Kinet. , 2017, 49: 464-476 doi: 10.1002/kin.21090
[3]
Wang Y, Wang H, Wang X, Xiao Y, Zhou Y, Su X, Cai J, Sun F. Sci. Total Environ. , 2020, 730: 139034 doi: 10.1016/j.scitotenv.2020.139034
[4]
Shen H, Xue W, Fu F, Sun J, Zhen Y, Wang D, Shao B, Tang J. Chem. Eur. J. , 2018, 24: 18463-18478 doi: 10.1002/chem.201804267
[5]
Shen J, Wang R, Liu Q Q, Yang X F, Tang H, Yang J. Chin. J. Catal. , 2019, 40: 380-389 doi: 10.1016/S1872-2067(18)63166-3
[6]
Shinde S L, Nanda K K. ACS Sustainable Chem. Eng. , 2019, 7: 66116618
[7]
Uribe Arizmendi I, Anducho Reyes M A, Ramirez Vargas M R, Cadena-Ramirez A, Muro-Urista C R, Tellez-Jurado A. Water Air Soil Pollut. , 2020, 231: 307 doi: 10.1007/s11270-020-04705-9
Atta A M, Moustafa Y M, Al-Lohedan H A, Ezzat A O, Hashem A I. ACS Omega, 2020, 5: 2829-2842 doi: 10.1021/acsomega.9b03610
[10]
Khataee A R, Pons M N, Zahraa O. J. Hazard. Mater. , 2009, 168: 451457
[11]
Manjari G, Saran S, Radhakrishanan S, Rameshkumar P, Pandikumar A, Devipriya S P. J. Environ. Manage. , 2020, 262: 110282 doi: 10.1016/j.jenvman.2020.110282
[12]
El Nemr A, Hassaan M A, Madkour F F. Environ. Process Int. J. , 2018, 5: 95-113 doi: 10.1007/s40710-018-0284-9
[13]
Mahamuni N N, Adewuyi Y G. Ultrason. Sonochem. , 2010, 17: 9901003
[14]
Paixao K, Abreu E, Samanamud G R L, Franca A B, Loures C C A, Baston E P, Naves L L R, Bosch J C, Naves F L. J. Environ. Chem. Eng. , 2019, 7: 102801 doi: 10.1016/j.jece.2018.11.045
Thomas S, Sreekanth R, Sijumon V A, Aravind U K, Aravindakumar C T. Chem. Eng. J. , 2014, 244: 473-482 doi: 10.1016/j.cej.2014.01.037
[19]
Atarod M, Nasrollahzadeh M, Sajadi S M. J. Colloid Interface Sci. , 2016, 462: 272-279 doi: 10.1016/j.jcis.2015.09.073
[20]
Wu J M, Wen W. Environ. Sci. Technol. , 2010, 44: 9123-9127 doi: 10.1021/es1027234
[21]
Chen H, Motuzas J, Martens W, da Costa J C D. Appl. Catal. B, 2018, 221: 691-700 doi: 10.1016/j.apcatb.2017.09.056
[22]
Li R, Zhu X, Yan X, Kobayashi H, Yoshida S, Chen W, Du L, Qian K, Wu B, Zou S, Lu L, Yi W, Zhou Y, Fan J. ACS Catal. , 2017, 7: 1478-1484 doi: 10.1021/acscatal.6b03370
[23]
Chen S, Liang S P, Wu B L, Lan Z H, Guo Z W, Kobayashi H, Yan X Q, Li R. ACS Appl. Mater. Interfaces, 2019, 11: 33946-33954 doi: 10.1021/acsami.9b11023
[24]
Zhu X, Du L, Guo Z, Chen S, Wu B, Liu X, Yan X, Takeuchi N, Kobayashi H, Li R. Catal. Sci. Technol. , 2018, 8: 6217-6227 doi: 10.1039/C8CY01551A
[25]
Meitzner G. In Situ Spectroscopy in Heterogeneous Catalysis. Haw J F. ed., Weinheim: Wiley VCH Verlag GmbH, 2002: 179-194
[26]
Xu F, Meng K, Cheng B, Yu J, Ho W. ChemCatChem, 2018, 11: 465472
[27]
Gonchar A, Risse T, Freund H J, Giordano L, Di Valentin C, Pacchioni G. Angew. Chem. Int. Ed. , 2011, 50: 2635-2638 doi: 10.1002/anie.201005729
[28]
Pirozzi D, Imparato C, D'errico G, Vitiello G, Aronne A, Sannino F. J. Hazard. Mater. , 2020, 387: 121716 doi: 10.1016/j.jhazmat.2019.121716
[29]
Zhang C W, Li T Y, Zhang J Y, Yan S, Qin C Y. Appl. Catal. B, 2019, 259: 118030 doi: 10.1016/j.apcatb.2019.118030
Figure 4
UV-Vis absorption spectra of AR1 dyes degraded at room temperature by catalysts with different loaded metals:
(a) Ag/MgO, (b) Au/MgO, (c) Pd/MgO, (d) Pt/MgO; (e) Pictures of degraded AR1 solution corresponding to (a)


 下载:
下载:








 下载:
下载:













 下载:
下载:
