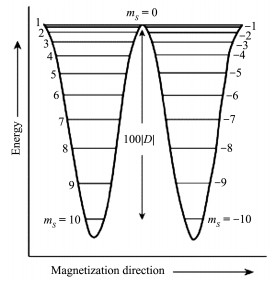
图 1.
[Mn12]-OAc的不同mS态的势能图
Figure 1.
Plot of potential energy as a function of mS quantum number for [Mn12]-OAc

单分子磁体(single molecule magnets,SMMs)是一类由顺磁性金属离子与适当配体组装而成且具有慢弛豫行为的配合物。在外磁场下对这类配合物磁化后,再降低温度至其阻塞温度(TB)下并撤去外磁场,配合物的磁矩能得以保持。因此,这类配合物在低于其TB时也会表现出经典磁体所具有的磁滞回线现象。然而,与经典磁体不同的是,有些单分子磁体的磁滞回线上还会出现由量子隧穿效应所导致的小台阶。一般来讲,单分子磁体为多核金属配合物,而近年来报道的单核3d或4f SMMs称为单离子磁体(single ion magnets,SIMs)[1]。值得一提的是,除少数结构单元距离较远的三维配合物表现出单分子磁体行为外,一般所指的单分子磁体为零维结构配合物。因此,在合成单分子磁体时,人们常选择一些多齿螯合配体,尤其是有机多齿螯合配体来组装单分子磁体配合物。自从1993年发现[Mn12]- OAc配合物表现出单分子磁体性质以来[2],设计合成更高能垒和阻塞温度的SMMs一直受到国内外合成化学家、材料学家和物理学家的关注,原因在于这类材料在高密度信息存储设备、量子计算及自旋电子学等方面存在潜在应用[3-4]。
配合物具有单分子磁体性质的原因在于这类分子本身存在一种能阻碍分子磁矩翻转的能垒。对于多核3d过渡金属配合物(最早引起关注的一类单分子磁体),配合物内3d金属离子的d轨道角动量在配位场下发生淬灭,因此,配合物分子只能用自旋磁矩表示,其大小为mS×μB(μB为玻尔磁子,mS为配合物的基态自旋S所对应的自旋磁量子数)。如果采取适当近似,则可只考虑二阶晶体场效应。因此,表达这类配合物能垒的哈密顿算符可用式(1)表示:
|
$ \hat H = D[S_z^2 - S(S + 1)/3{\rm{ }} + g{\mu _\rm{B}}{H_z}{S_z}] $ |
(1) |
其中,Sz为自旋投影算符,Hz表示z方向外加磁场强度,D为磁各向异性参数(用零场分裂参数表示,它可解除基态自旋S多重度的简并态。如不考虑D,S所对应的(2S+1)个mS的能量是相同的)。(1)式中基态自旋S所对应的不同的mS态的能量可通过(2)式很容易计算出来:
|
$ E(m_S) = D[m_S^2 - S(S + 1)/3] + g{\mu _\rm{B}}{m_S}{H_z} $ |
(2) |
其中,-S≤mS≤S。
利用公式(2),人们便可画出多核3d过渡金属单分子磁体的不同mS态的能级图。例如,对于[Mn12]-OAc,其基态自旋S=10;把S代入(2)式可得到(3)式:
|
$ E({m_S}) = D(m_{_S}^2 - 110/3) + g{\mu _\rm{B}}{m_S}{H_z} $ |
(3) |
其中,-S≤mS≤S。
[Mn12]-OAc的能级图如图 1所示。当D为负值时,配合物磁矩mS发生翻转,需要从一种磁矩方向(如mS=S)一步步地跨过比mS小1的态,直到通过|mS|最小的态,然后再一步步地下降到mS=-S态。此外,这些态之间跨越之所以能实现,主要得益于“自旋-声子”耦合作用,而此耦合作用可看成是由晶格振动所导致的晶体场微扰产生的。对于多核过渡金属SMMs,弛豫过程一般满足阿伦尼乌斯公式:
|
$ \tau = {\tau _0}\exp [U/(kT)] $ |
(4) |
其中U为能垒,它表示mS=S态与|mS|最小的态之间的能量差,其大小为U=(S2-1/4)|D|(S为半整数)或U= S2|D|(S为整数)[5-8]。
大约在1993—2008年期间,人们集中于多核3d过渡金属配合物的研究,试图通过增加配合物的核数来提高它们的基态自旋S及磁各向异性参数D的数值。在此期间,人们合成了大量不同核数的Mn、Fe、Co、Ni、Cu配合物[5-6],其中以含MnⅢ的高核Mn和含FeⅢ的高核Fe配合物居多。有意思的是,一些结构新颖、核数较高的配合物在此期间被报道出来,如[Mn25][7-8]、[Mn84][9]、[Mn31][10]、[Mn10][11]、[Mn19][12]等,且其中一些配合物的S确实较高,如[Mn25]的基态自旋S达到51/2或61/2[7-8]。然而,在这近十五年时间里(1993—2008年),只有[Mn6]配合物的能垒稍高于最初报道的[Mn12]-OAc配合物[13]。进一步研究发现,这些高自旋配合物的D值几乎为0[7, 11]。这主要由于这些配合物的对称性一般也较高,如[Mn10]分子结构为T对称性,从而导致MnⅢ的Jahn-Teller轴相互垂直并进而抵消了配合物内每个MnⅢ的磁各向异性[11]。此外,在2007—2011年期间,许多课题组开始反思纯多核过渡金属SMMs的能垒的调控方式并对S和D之间的关系进行理论研究。如2007年,Waldmann指出能垒不随着S的增大而增加,并指出应该通过提高磁各向异性来提高单分子磁体性能[14]。2008年,Ruiz等也认为配合物能垒和基态自旋S不呈线性关系的增长,高的S会导致低的D值[15]。2011年,Neese等同样认为增加配合物核数和基态自旋并不是提高配合物能垒的有效手段,而认为应该从小分子甚至单核配合物入手研究控制能垒的影响因素,并提出用这些小分子作为单元组装成高能垒配合物[16]。总之,自2008年后,人们已经把研究目光从多核纯过渡金属单分子磁体的研究转向单核3d、单(多)核4f及3d-4f单分子磁体的研究。
3d-4f单分子磁体一般是指顺磁性3d过渡金属离子与顺磁性4f金属离子通过适当配体所构建的且具有单分子磁体性能的混金属配合物。目前人们研究3d-4f单分子磁体主要基于2个目的:一是向多核3d配合物内引入磁各向异性较大的稀土离子以得到比纯过渡金属配合物能垒更高的单分子磁体配合物;二是通过调控3d-4f配合物内顺磁离子间磁耦合作用以调控3d-4f配合物的磁弛豫行为。需要指出的是,3d-4f单分子磁体中4f离子较强的旋轨耦合作用使得稀土离子的电子结构应该用总量子数J描述,这意味着对于4f稀土离子甚至3d-4f配合物,S不再是适用量子数。因此,在描述3d-4f单分子磁体的能垒时不能像多核过渡金属配合物那样用U=(S2-1/4)|D|或S2|D|描述。第一例具有单分子磁体性质的3d-4f单分子磁体是日本科学家Kido等报道的[CuTb]双核异金属配合物[17]。然而,3d-4f配合物的研究直到2008年左右才逐渐引起人们的重视。人们选择的3d离子主要为顺磁性CrⅢ、MnⅢ、FeⅢ、CoⅡ、NiⅡ和CuⅡ离子,而选择的4f离子一般为磁各向异性较大的DyⅢ、TbⅢ、HoⅢ及ErⅢ等。此外,近年来,为了降低或消除3d-4f配合物中弱的磁耦合作用及研究配合物弛豫行为与3d离子关系,人们也选择第一过渡系中的抗磁性离子如CoⅢ和ZnⅡ离子等,并报道了大量的CoⅢ-Dy和ZnⅡ-Dy配合物。值得一提的是,含DyⅢ离子的3d-Dy单分子磁体在3d-4f中数目最多、能垒和阻塞温度一般最高,且引起人们重视的程度也最多。基于以上考虑,我们在本综述中分别讨论了第一过渡系中顺磁离子和抗磁离子与DyⅢ形成的单分子磁体的研究情况,并对该领域进行展望和提出进一步提高它们的能垒和阻塞温度的思路。
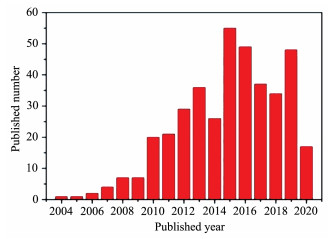
使用“single molecule magnet”、“3d”和“Dy”为关键词在web of science进行搜索,粗略的文献统计如图 2所示(ZnⅡ-DyⅢ和CoⅢ-DyⅢ单分子磁体也添加进统计里),可以发现第一过渡系金属-镝单分子磁体报道的文献数在2003—2015年期间逐年上升,然而2015年后报道的数量有下降趋势,并且2015年后报道的大多数配合物的3d离子一般为CoⅢ和ZnⅡ离子。此外,进一步文献调研发现,最近四年以来,除第一过渡系金属-镝单分子磁体数量减少,多核Dy及纯多核过渡金属单分子磁体的论文数量也在逐年减少,而单核稀土(以Dy居多)单分子磁体(又称单离子磁体)的论文数量在逐年增多。这主要源于Dy基单离子磁体中Dy配位几何相对较易调控,并报道了数例高能垒和高阻塞温度的配合物[18]。作者认为含DyⅢ的多核配合物(包括纯Dy和TM-Dy)的能垒和阻塞温度难以调高,除了与配合物内顺磁性金属离子之间弱的磁耦合作用有关外,还与配合物内DyⅢ的配位几何的对称性及不同DyⅢ的磁各向异性轴的方向难以控制有关。下面作者对TM-Dy基单分子磁体配合物近年来的研究情况和研究趋势进行综述,并提出进一步提高该类配合物单分子磁体性能的方法,期望能给该领域研究提供指引作用。如前所述,我们在本综述中分别对顺磁性3d离子与DyⅢ构筑的SMMs和第一过渡系抗磁性离子与DyⅢ构筑的SMMs的研究情况进行概述。需要指出的是,我们仅重点讨论具有新颖结构和/或具有优异单分子磁体性能的配合物,其他代表性的TM-Dy单分子磁体列于表S1~S6(Supporting information)。
在研究顺磁性3d离子与DyⅢ所构建的混金属单分子磁体(3d-Dy SMMs)的早期,该领域研究目的是解决部分高自旋高对称的纯3d过渡金属配合物的磁各向异性较低的问题,希望通过加入磁各向异性较大的稀土离子如DyⅢ来提高配合物的磁各向异性,进而提高能垒。需要指出的是,DyⅢ的基态光谱项6H15/2在完美的单轴各向异性晶体场下会分裂出8个双重简并的mJ(mJ=±15/2、±13/2,±11/2、±7/2、±5/2、±3/2、±1/2),这些双重简并的微观态只有在外磁场下才会进一步分裂。由于DyⅢ离子的基态光谱项(6H15/2)与第一激发态(6H13/2)或更高激发态有几百甚至上千cm-1的能级差,因此DyⅢ常用于合成单分子磁体或单离子磁体。实际上,对于大多数3d-Dy配合物,其双稳态往往由不同的mJ态线性组合而成,这会降低各个态的磁各向异性,进而降低单分子磁体能垒。因此,提高3d-Dy单分子磁体关键是尽量使每个双稳态对应某个mJ态,尽量减少不同mJ态的混杂。另外,3d-Dy配合物内弱的磁耦合作用会降低基态和低的激发态能级差,进而降低能垒;反之,强的磁耦合作用不仅可提高能垒,还会增大磁滞回线的矫顽场和面积。因此,提高3d-Dy内磁耦合作用需要引起更多的关注。
CrⅢ离子3d轨道上有3个电子,在八面体配位场下,其d电子排布为t2g3eg0,理论上,该离子不会有Jahn-Teller效应,从而使得该离子磁各向异性较弱。因此,CrⅢ离子在3d-4f配合物研究初期并没有引起太多重视。然而,近年来人们对Cr-Dy基单分子磁体产生较大的兴趣,原因在于相比于其他顺磁性3d金属离子,CrⅢ与DyⅢ间磁耦合作用较大(可达到|J|= 8~10 cm-1),导致磁滞回线的阻塞温度较高(可达到3.7 K),且有较高的矫顽场(1.8 K时可达到2.7 T)和磁滞回线面积。目前报道的具有优异单分子磁体性能且进行详细研究的主要有[Cr4Dy4]和[Cr2Dy2]两类拓扑结构的配合物。
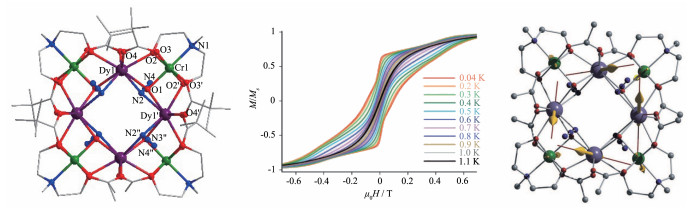
第一类Cr-Dy单分子磁体为[Cr4Dy4]配合物。[Cr4Dy4(μ3-OH)4(μ3-N3)4(mdea)4(piv)4]·3CH2Cl2 (1)[19]最初由Powell等于2010年报道,这也是第一个具有优异单分子磁体性能的配合物。这个配合物的4个DyⅢ离子通过4个μ3-OH-和4个端基桥联的N3-连接,形成完美的正方形,DyⅢ …DyⅢ之间的距离为0.403 39(2) nm。处在每对DyⅢ之间的μ3-OH-又桥联一个CrⅢ离子,CrⅢ…DyⅢ之间的距离为0.333 33(4) nm(图 3左)。交流磁化率研究表明,该配合物的有效能垒(Ueff)为15 K,弛豫时间(τ0)为1.9(1)×10-7 s;通过micro-SQUID测量显示(图 3中),温度达到1.1 K时可观察到磁滞回线。此外,理论计算研究表明DyⅢ- CrⅢ之间磁耦合作用设定为较大值(-4.5 cm-1)时,可很好地拟合χMT-T和M-H曲线,且4个DyⅢ的磁各向异性主轴几乎垂直于Dy4平面(图 3右)。值得一提的是,这类配合物单分子磁体性能与DyⅢ之间的桥联配体有关。如把上述配合物的4个μ3-OH-和4个N3-换成2.75个μ3-OH-、1.25个μ3-OCH3及4个F-,则其Ueff可升高到55 cm-1(τ0=6.1×10-8 s),且温度在3.5 K时也可观测到磁滞回线[20]。此类单分子磁体稍显不足的是它们的磁滞回线的矫顽场较小。
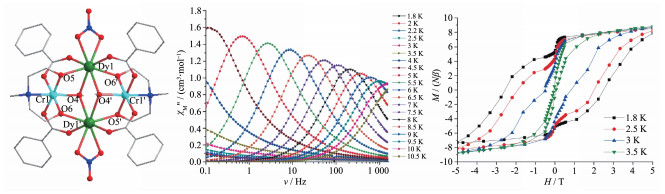
第二类Cr-Dy单分子磁体为[Cr2Dy2]配合物。最初研究的2个该类型配合物由Bendix等于2012年报道,其分子式为[Cr2Dy2(μ-F)4(μ-OH)2(py)4(hfac)6] (2)和[Cr2Dy2(μ-F)4F2(py)6(hfac)6] (3)[21]。配合物2中2个Cr和2个Dy形成一个处在同一平面的平行四边形结构,距离较短的顶点被2个Cr占据,它们通过2个μ-OH-连接,然后每个Cr与处于平行四边形两端的DyⅢ通过4个μ-F-连接。而配合物3中2个Cr和2个Dy形成处在同一平面的菱形顶点上。与2不同的是,配合物3中2个Cr没有配体连接,而是菱形的4个边通过4个μ-F-连接。交流磁化率研究表明,配合物2有场致频率依赖现象,Ueff和τ0分别为4.59(18) cm-1和2.8×10-5 s;而配合物3在加场下也只有微弱的χ″信号,没有峰值。这与实验拟合的Cr-Gd之间较弱的磁耦合作用有关(配合物2:JCr-Gd= 0.43(5) cm-1,配合物3:JCr-Gd=0.57(7) cm-1)。磁耦合作用较弱是由于Cr与Dy之间通过F-桥联导致的。如前所述,Cr与Dy之间可产生较强的磁耦合,为了调控磁耦合作用,人们尝试使用含O原子的配体桥联。随后,在2013年,Murray等报道了一个“蝴蝶”型配合物[CrⅢ2DyⅢ2(OMe)2(O2CPh)4(mdea)2(NO3)2] (4,mdeaH2 =N-甲基二乙醇胺)[22],其中2个Dy处在“蝴蝶”身体位置,而2个Cr处在“蝴蝶”翼的位置。2个Dy通过2个μ-OMe-连接,Dy与Cr之间通过N-甲基二乙醇胺的羟基O和苯甲酸的2个羧基O连接(图 4左)。磁性研究表明配合物4具有单分子磁体行为(图 4中),其Ueff和τ0分别为(77±0.5) K和5.1×10-8 s。ZFC-FC曲线表明,该配合物的TB为3.7 K;micro-SQUID测试表明(图 4右),该配合物到3.5 K时仍能显示磁滞回线。尤其重要的是,该配合物有较大的矫顽场,达到2.8 T。理论计算研究表明,该配合物中Dy具有较高的轴向磁各向异性,第一激发态能级大于100 cm-1,且Cr和Dy之间磁耦合作用较大(>15 cm-1),这些计算结果很好地说明了该配合物具有优异的单分子磁体行为的原因。此外,Murray等用六氟乙酰丙酮取代上述配合物中的苯甲酸[23],然而Ueff却降低到29 cm-1,且能观察到磁滞回线的温度降为2.2 K,矫顽场大约为0.25 T。这可能与Cr-Dy间磁耦合作用和Dy的配位几何对称性降低及量子隧穿效应增大有关。此外,当把苯甲酸替换成2-氯-4,5-二氟苯甲酸或对叔丁基苯甲酸时,能垒分别为61 cm-1(τ0=2.1×10-7 s)和45 cm-1(τ0=7.7×10-8 s),可观察到磁滞回线的温度分别为4.4和3.1 K,矫顽场分别为4.4和2.2 T[24]。这些参数都表明,使用推电子基团的苯甲酸(对叔丁基苯甲酸)不利于[Cr2Dy2]单分子磁体性能提高,而使用吸电子基苯甲酸配体(2-氯-4,5-二氟苯甲酸)有利于提高该类分子的SMM性能。总之,该类单分子磁体优点是有较大的矫顽场和磁滞回线面积。
此外,[Cr2Dy4]、[CrDy2]、[CrDy6]、[Cr6Dy6]等不同拓扑结构的Cr-Dy配合物也已被报道[25-28]。这些配合物也具有频率依赖现象和较高的能垒,但没有观察到磁滞回线或磁滞回线的矫顽场较低。如Murray等报道了一个[CrDy6]配合物[CrⅢDy6Ⅲ(OH)8 (ortho-tol)12(NO3)(MeOH)5]·3MeOH (5,ortho-tol=ortho-toluate)[27],其中每3个DyⅢ离子通过一个μ3-OH-离子连接形成一个三角形,然后这2个三角形通过6个μ3 -OH-与处于2个三角形之间的CrⅢ连接。因此,该结构可看成由2个共顶点的三角锥构成。每个Dy3三角形中,Dy…Dy之间距离为0.374 9~0.378 0 nm,2个三角形平面质心之间距离为0.538 nm。通过SHAPE软件计算表明,配合物5中每个DyⅢ采取八配位三角十二面体几何构型,但计算出的CShM (continuous shape measures)参数较大(1.2~2.7)。需要说明的是,CShM值越接近于0,Dy的配位几何越接近理想多面体。因此,配合物5中的DyⅢ的配位几何偏离理想多面体较大。密度泛函理论(DFT)计算表明,配合物5内Dy…Cr和Dy…Dy的磁耦合作用(计算时把Dy换成Gd)均较小,分别为-0.08和-0.043 cm-1。Micro-SQUID研究表明,配合物5具有单分子磁体和环形磁矩双重特征。
Mn-Dy单分子磁体是3d-Dy配合物中研究较早的一类,并且许多结构和磁性奇异的Mn-Dy配合物已被报道。2006年Powell课题组报道了一例[Mn19]配合物[12][MnⅢ12MnⅡ7(μ4-O)8(μ3-N3)8(HL)12(MeCN)6]Cl2· 10MeOH·MeCN (6,H3L=2,6-二(羟甲基)-4-甲基苯酚),它的基态自旋S达到83/2。然而,该配合物对称性较高,使得[Mn19]配合物内MnⅢ离子的磁各向异性相互抵消,进而使得配合物的磁各向异性为一个极小的正值。结构研究发现,[Mn19]配合物中心有一个MnⅡ。如果使用稀土离子(如DyⅢ)替换MnⅡ,则相应配合物的磁各向异性将会提高。为此,该课题组于2009年报道了Mn18Dy配合物,[MnⅢ12MnⅡ6DyⅢ(μ4-O)8 (μ3-Cl)6.5(μ3-N3)1.5(HL)12(MeOH)6]Cl3·25MeOH (7,图 5左)[29]。磁性研究发现,配合物7具有单分子磁体性质,且在温度低于0.5 K时,可观察到磁滞回线。
2015年,唐金魁课题组报道了一系列较为少见的线型(LnⅢ-MnⅡ-LnⅢ)配合物[Ln2Mn(C7H5O2)8](图 5中,Ln=Gd (8)、Tb (9)、Dy (10)、Ho (11)、Er (12))。通过对配合物8磁性拟合发现,MnⅡ…GdⅢ和GdⅢ…GdⅢ之间均为弱反铁磁耦合,分别为-0.35(2)和-0.11(3) cm-1。此外,配合物9和10表现出慢弛豫现象,说明这2个配合物具有单分子磁体行为。更重要的是,10还表现出了较高的能垒(Ueff=92.4 K)和双缓慢弛豫过程,其中高温区域和低温区域的弛豫过程可能分别来源于配合物内DyⅢ的单离子磁行为和MnⅡ… DyⅢ之间的弱的磁耦合作用[30]。2016年Agapie课题组通过氧化还原的化学方法调控合成了2类立方烷形Mn-Dy簇合物:[DyMnⅣ3O4] (13)和[DyMnⅣ2MnⅢO4] (14),并研究了锰氧化态对单分子磁体性能的影响。研究发现,配合物13显示单分子磁体的特性。理论上,配合物14含有磁各向异性的MnⅢ离子而使之应具有更加优异的单分子磁体特性,然而,令人意外的是,14却没有表现这一性质。通过对Y-Mn的类似物磁性拟合发现,配合物13中顺磁离子间磁耦合作用要强于14,这说明配合物13中MnⅣ3单元的铁磁耦合作用和Dy-Mn间的耦合作用共同使之具有更优异单分子磁体特性[31]。
同年,我们报道了一例[Mn8Dy2]配合物[Mn8Dy2O2(OH)2{(py)2CO2}4(teaH)4(CH3COO)6] (15)。处于配合物15中心的Dy1、Dy1a、Mn4、Mn4a组成了一个“蝴蝶”型的拓扑结构,2个MnⅢ作为“蝴蝶”的身体,2个DyⅢ作为“蝴蝶”的翅膀,而其余的6个锰离子坐落在“蝴蝶”的两侧,通过μ3-O2-离子桥联起来(图 5右)。结构的有趣之处在于DyⅢ的配位点被6个O原子和2个N原子占据,而没有和溶剂分子和小的阴离子基团发生配位,这在3d-4f配合物中是少见的,而交流磁化率显示此配合物具有较小的能垒,我们推测可能是由于DyⅢ离子的低对称性的配位构型导致的[32]。
2018年,Powell课题组使用不同配体、不同Mn盐和不同溶剂获得了3个系列共22个Mn-Ln配合物,分别为:[MnⅢ2MnⅡ2Ln2(μ4-O)2(H2edte)2(piv)6(NO3)2] (16)、[MnⅢ2MnⅡ2Ln2(μ4-O)2(edteH2)2(benz)6(NO3)2] (17)和[MnⅢ2MnⅡ2Ln2(μ4-O)2(edteH2)2(piv)8] ·solv (18) (Ln= La、Ce、Pr、Nd、Eu、Sm、Gd、Tb、Dy、Ho、Er等;H2edte= N,N,N′,N′-tetrakis(2-hydroxyethyl)ethylenediamine,piv=pivalate,benz=benzoate;solv=MeCN和/或tolu- ene)。这3个系列配合物均以[MnⅡ2MnⅢ2LnⅢ2(μ4-O)2 (μ3-O)4]8+为核心(图 6左),它们都是由不同配体上的氧原子桥联成的共面双立方烷结构。直流磁化率研究表明MnⅢ…MnⅢ是反铁磁耦合,而MnⅡ…MnⅢ、MnⅡ…LnⅢ、MnⅢ…LnⅢ是弱的铁磁耦合。在这22个配合物中,含Sm、Tb、Dy、Ho的配合物具有单分子磁体行为。该研究表明在不改变核结构时,通过使用不同配体可达到调控单分子磁体磁弛豫行为的目的[33]。
同年,Rajaraman等也报道了一类Mn3Ln2配合物[MnⅣMnⅢ2LnⅢ2O2(RCO2)4(mdea)3(NO3)2(MeOH)](图 6中,Ln=Dy (19)、Tb (20)、Gd (21和26)、Eu (22和27)、Sm (23)、Nd (24)、Pr (25);RCO2-=苯甲酸酯(19~25)或者甲苯甲酸酯(26和27);mdeaH2=N-甲基二乙醇胺),其中19和25的交流磁化率虚部表现出频率依赖现象,说明其可能有单分子磁体行为。从头计算表明配合物19和25的零场单分子磁体行为的来源并不仅限于单个离子的各向异性,利用POLY_ANISO程序,发现单分子磁体行为是由MnⅢ/Ⅳ-LnⅢ和LnⅢ-LnⅢ间弱的耦合作用导致的。然而,它们之间的弱的耦合作用仅能淬灭小的或中等的量子隧穿效应,处在激发态的大的量子隧穿效应不能有效淬灭,从而使得激发态仅仅位于基态上方几个波数处,进而导致能垒很小(图 6右)。此外,MnⅢ离子的各向异性的存在导致了配合物22、26和27在外加场的作用下也具有单分子磁体行为,这为未来改善单分子磁体的性质提供了参考[34]。
在Mn-Dy配合物发展过程中也报道了其他有趣的结构,如环状的[Mn4Ln4]、[Mn8Ln8]配合物[35-36]、具有多弛豫过程且叠氮根数目高达12个的[Mn4Dy2]配合物、杆状的[Mn6Ln2]配合物等[37-38]。
常见的Fe-Dy配合物中Fe的价态为+3。FeⅢ离子在配位场下往往表现出高自旋(S=5/2)。因此,使用高自旋态的FeⅢ所合成出的Fe-Dy配合物可能具有优秀的单分子磁体行为。然而,目前报道的Fe-Dy单分子磁体大约仅有十多个,相对于其他3d离子与Dy构建的单分子磁体,数目不是很多。主要有2个原因:(1)高自旋FeⅢ磁各向异性较低;(2) FeⅢ与DyⅢ之间磁耦合作用较小。值得一提的是,文献中也有极少量的FeⅡ和DyⅢ构成的单分子磁体配合物。下面我们介绍几例Fe-Dy单分子磁体。
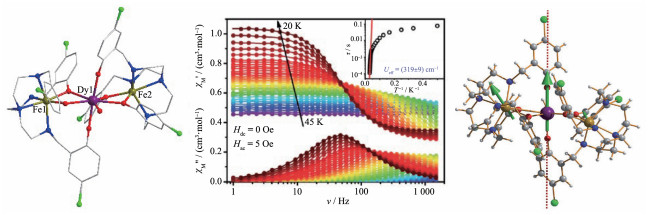
最具有代表性的Fe-Dy单分子磁体是2014年童明良课题组[39]报道的一例线性三核配合物[Fe2Dy(L)2 (H2O)]ClO4·2H2O (28,图 7左,L=2,2′,2″-(((nitrilotris (ethane-2,1-diyl))tris(azanediyl))tris(methylene))tris(4-chlorophenol))。在此配合物中,DyⅢ离子的晶体场为五角双锥型(准D5h对称),2个不对称的FeⅡ离子则分别采取Oh对称性的八面体构型和C3v对称性的三棱柱构型,FeⅡ和DyⅢ之间通过配体的酚氧桥联。磁性研究表明,配合物28具有单分子磁体行为(图 7中),其能垒高达319 cm-1(459 K),是当时3d-4f单分子磁体中能垒最高的。大量研究表明,具有D5h配位对称性的含Dy配合物往往具有优异的单分子磁体性能。因此,28具有优秀的磁弛豫行为可能源于其中DyⅢ的五角双锥的配位几何。此外,通过从头计算发现,低占据激发态也具有较强的轴向性质(图 7右),导致28的量子隧穿效应得到了有效的抑制[39]。
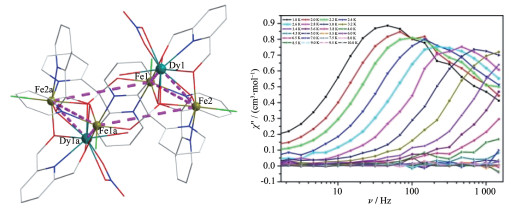
2016年,Powell课题组合成了一例[Fe4Dy2]环[Fe4Dy2(μ3-OH)2(nbdea)4(C6H5CO2)8]·MeCN (29),并研究了中心金属Fe和Dy的磁耦合作用。磁性研究表明,该配合物在低温下表现为铁磁相互作用,并表现出单分子磁体特性,且相似构型的[Ga4Dy2]环配合物拥有与[Fe4Dy2]相近的能垒,但弛豫时间要更长,这表明顺磁性的FeⅢ离子代替抗磁性的GaⅢ离子可以有效地抑制量子隧穿效应[40]。2017年,Powell组又合成了一系列的[Fe4Ln2]配合物(Ln=Y、Gd、Tb、Dy、Ho、Er),其中Dy、Tb、Ho表现出了单分子磁体行为,通过低温下的高场/高频电子顺磁共振测量[Fe4Ln2]配合物中3d离子的各向异性和3d与4f离子间的交换相互作用,发现随着Ln离子原子序数的增加,配合物的JFe-Ln铁磁交换相互作用将会减少,从JFe-Ln与有效能垒的一致性关系来看,3d和4f离子间的交换相互作用应该是增强3d-4f单分子磁体的性质的一个重要因素[41]。
2019年,我们使用2,2-二吡啶酮和2,6-吡啶二甲醇得到一系列新颖结构配合物Fe4Dy2{(py)2CO2}4 (pdm)2(NO3)2(H2O)2Cl4] ·CH3CN·H2O (30,Ln=DyⅢ;(py)2CO2H2=the gem-diol form of di-2-pyridyl ketone,pdmH2=2,6-pyridinedimethanol)[4]。配合物中金属离子构成“椅式”形状,4个FeⅢ占据“椅”的主体,2个DyⅢ占据“椅”的两端(图 8左)。有意思的是,[Fe4Dy2]配合物具有单分子磁体行为(图 8右),使用τ-1=CTn+τ0-1exp[-Ueff/(kBT)]拟合,得到C=1.6(2) ×10-10 s-1·K-13,τ0=4.7(1)×10-7 s及Ueff=19.3(8) K。通过拟合[Fe4Gd2]的类似物的直流磁化率数据发现,FeⅢ与GdⅢ之间有较强磁耦合作用(分别为-6.0(1)和2.8(1) cm-1),且对[Fe4Dy2]的理论计算也表明磁耦合作用较强。这可能是配合物30具有单分子磁体性质的原因。
CoⅡ离子在八面体配位场下电子排布一般采取t2g5eg2形式,有较强的旋轨耦合导致的磁各向异性,其在3d顺磁过渡金属离子中磁各向异性也是较大的。因此,在3d-4f研究起始阶段,人们热衷于合成CoⅡ-Dy配合物。然而,相比于其他3d离子和Dy构建的单分子磁体,CoⅡ-Dy单分子磁体数目同样较少,大约不到20个。由于CoⅡ在碱性溶液中易变成抗磁性CoⅢ离子。通过适当条件合成的CoⅢ-Dy配合物可消除3d离子与DyⅢ的磁耦合作用,CoⅢ离子在相应配合物中起到磁稀释作用,可提高相应配合物能垒和阻塞温度。因此,最近五年,CoⅢ-Dy单分子磁体研究引起人们重视。为便于讨论,我们在后一部分专门讨论CoⅢ-Dy单分子磁体。
2017年,唐金魁等利用p-tert-butylcalix[8]arene (H8C8A)配体在甲醇和N,N-二乙基甲酰胺(DEF)溶剂里得到了[CoⅡ4DyⅢ4]配合物[(DyⅢ4CoⅡ4)(C8A)2(O)2 (DEF)8(H2O)4]·0.5CH3OH (31),它的[CoⅡ4DyⅢ4]核夹在2个对端的芳烃配体之间(图 9左),其中2个DyⅢ采取八配位反四棱柱构型,而另外2个DyⅢ采取七配位单帽八面体构型。有意思的是,4个CoⅡ离子配位数和空间构型也不同,2个CoⅡ采取五配位扭曲的三角双锥构型,而另外2个CoⅡ为六配位扭曲八面体构型。交流磁化率研究表明31在0 Oe外加直流场下有较强频率依赖现象,且在温度低于4.5 K时χ″有最大值,表明其可能具有单分子磁体行为。此外,Cole -Cole图接近于半圆形,对其通过德拜模型拟合得到的α值在0.1~0.41之间,表明31有相对较宽的弛豫时间分布。进一步研究31的lnτ对T-1曲线发现该配合物存在Raman和Orbach两种过程,有效能垒为10.38 K[42]。
2017年,宋友等报道了3个配合物[Dy2ZnⅡ2(L)4 (NO3)2(CH3OH)2] (32)、[Dy2MnⅡ2(L)4(NO3)2(DMF)2] (33)和[Dy2CoⅡ2(L)4(NO3)2(DMF)2] ·2DMF (34) (H2L= (E)-2-ethoxy-6-(((2-hydroxyphenyl)imino)methyl)phenol)。3个配合物的金属核均采用常见的“蝴蝶”型结构,过渡金属离子处在“蝴蝶身体”上,而DyⅢ离子处在“蝴蝶翼”的位置。交流磁化率研究表明,配合物32在0 Oe外加场下显示单分子磁体性质,且在低温处能观察到明显的磁量子隧穿效应(QTM),能垒为54.1 cm-1;32中的ZnⅡ换成CoⅡ后的配合物34也能显示单分子磁体行为,但在低温处没有明显QTM,能垒达到61.6 cm-1;而把32中的ZnⅡ换成MnⅡ后得到的配合物33,只有弱的频率依赖现象,即使在1 000 Oe的外加直流场下也没有出现明显变化。此外,通过理论计算发现,顺磁性离子与DyⅢ的磁耦合作用使得基态隧穿能隙极小(10-7~10-9 cm-1),说明配合物33和34的QTM确实较低。此外,33和34除Orbach过程外,还有其他弛豫过程,这可能是34单分子磁体性能较差的原因[43]。
同年,Powell等报道了2个CoⅡ - Dy配合物[CoⅡ2Dy2(L)4(NO3)2(MeOH)2] ·2CH2Cl2 (35)和[CoⅡ2Dy2(L)4(NO3)2(DMF)2]·2C2H6CO (36),并与他们之前报道的[CoⅡ2Dy2(L)4(NO3)2(THF)2]·4THF (37)进行结构和磁性比较。这3个配合物均为双缺口立方烷型结构(图 9中),主要区别在于35和36配合物中CoⅡ分别被MeOH和DMF配位,而配合物37中CoⅡ被THF分子配位。通过SHAPE软件计算发现,尽管3个配合物中CoⅡ离子均采用六配位八面体构型,但偏离理想八面体的CShM值不同(37、35和36的CShM值分别为1.60、1.78和2.04),此外,3个配合物中DyⅢ的配位空间构型也不同,配合物37和36中DyⅢ为三角十二面体,而配合物35中DyⅢ为双帽三棱柱构型。这表明,CoⅡ上被不同配位分子占据会导致配合物内CoⅡ和DyⅢ的配位构型出现差异。此外,这种配位空间差异进一步导致CoⅡ离子与DyⅢ磁耦合作用大小的不同,并进而影响弛豫行为。这3个配合物均有双弛豫现象。配合物37的能垒U1=15.8 K,U2= 118.1 K;配合物35的能垒U1=17.9 K,U2=104.8 K;而配合物36的能垒U1=17.5 K,U2=94.5 K。在CoⅡ-Dy单分子磁体中,双弛豫现象较常见,一般把U2归因于配合物内DyⅢ的单离子能垒,而把U1归因于CoⅡ- Dy之间的磁耦合作用。为了验证U1来源于CoⅡ-Dy磁耦合,作者还合成了[ZnⅡ2DyⅢ2]类似物,交流磁化率研究表明,该Zn-Dy类似物确实为单弛豫,能垒为140.4 K。因此,作者通过比较3个配合物U1,首次发现配位溶剂分子的取代可改变CoⅡ-Dy之间磁耦合,且会影响配合物弛豫行为[44]。
2019年,de Souza等使用自由基配体TlTrzNIT合成了一个配合物[CoⅡ(hfac)(TlTrzNIT)2][Dy(hfac)4] (38,hfac为六氟乙酰丙酮,TlTrzNIT为1-(mtolyl)-1H-1,2,3-triazole-4-(4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide)。在配合物38中,2个TlTrzNIT自由基和一个hfac-与CoⅡ配位形成[Co(hfac)(TlTrzNIT)2]+配离子,其中CoⅡ为六配位扭曲八面体构型。有意思的是,[Co(hfac)(TlTrzNIT)2]+有手性异构,即右手螺旋的Δ-[Co(hfac)(TlTrzNIT)2]+配离子和左手螺旋的Λ-[Co(hfac)(TlTrzNIT)2]+配离子。它们的带负电荷的配离子为[Dy(hfac)4]-,其中DyⅢ被4个hfac-上的8个O配位,其配位构型介于反四棱柱和双帽三棱柱之间。[Co(hfac)(TlTrzNIT)2]+和[Dy(hfac)4]-之间通过氢键向ac平面排布。磁性研究表明,配合物38在0 Oe外加场下交流磁化率实部和虚部无明显频率依赖现象,然而,在1 500 Oe外加场下,有双弛豫现象,可能由于配合物内存在2种非晶体学等效的DyⅢ离子。需要指出的是,其中一个弛豫过程出现在温度极低的区域,因此,只能对其中一个弛豫行为进行拟合,考虑Orbach过程和Raman过程,并把n设定为9,得到τ0=3×10-6 s,Ueff/kB=12.8 K及C=0.074 s-1· K-9 [45]。
2018年,童明良课题组利用H3TTTTCl配体(H3TTTTCl=2,2′,2″ - (((nitrilotris(ethane - 2, 1 - diyl))tris (azanediyl))tris(methylene))tris-(4-chlorophenol)),得到2个配合物[CoⅡ2Dy(TTTTCl)2(MeOH)]NO3·3MeOH (39)和[CoⅡ2Dy(TTTTCl)2(MeOH)] [CoⅢ (HTTTTCl)] (NO3)2· 2.5MeOH·2H2O (40)。配合物39和40中[CoⅡ2Dy(TTTTCl)2(MeOH)]+配离子分别均含有2个CoⅡ和1个DyⅢ,形成稍弯曲的线性三核结构(图 9右),CoⅡ采取六配位扭曲八面体构型,而DyⅢ采取七配位压缩五角双锥几何构型(相比于39,配合物40中[CoⅡ2Dy (TTTTCl)2(MeOH)]+的DyⅢ压缩程度更高且更接近于规则的五角双锥)。配合物40中抗磁性的[CoⅢ (HTTTTCl)]+之间通过氢键形成{[CoⅢ(HTTTTCl)]+}2二聚体形式。此外,{[CoⅢ(HTTTTCl)]+}2二聚体处在2个[CoⅡ2Dy(TTTTCl)2(MeOH)]+之间。磁性研究表明,配合物39和40的能垒分别达到401和536 K。该研究也表明,通过引入抗磁性配位金属离子可调控配合物单分子磁体性能[46]。
NiⅡ在一般配位场下具有由较大的零场分裂导致的高的磁各向异性。因此,合成NiⅡ-Dy单分子磁体也引起人们重视,到目前为止,报道了大约30个Ni-Dy单分子磁体配合物,但该类配合物能垒都不是很高。
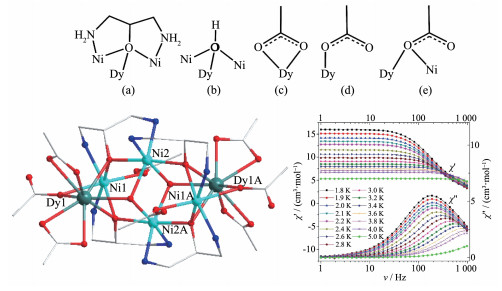
2013年,Müller等报道了一系列的[Ni2Ln3](Ln= Dy (41)、Gd (42)、Tb (43)、Ho (44))。X射线单晶衍射分析表明4个配合物为同构五核配合物,所有的Ln离子同Ni离子都是通过酚羟基O和醇羟基O原子桥联。NiⅡ离子配位几何为扭曲的平面四方形,如图 10左所示。配合物41中,3个DyⅢ均为八配位,其中Dy2采取扭曲的反四棱柱几何构型,而Dy1和Dy3采取扭曲的三角十二面体几何构型。磁化率测量表明配合物41中,分子内的NiⅡ离子与DyⅢ离子之间存在弱的铁磁耦合作用,而配合物42~44分子内的NiⅡ离子与LnⅢ离子之间存在弱的反铁磁耦合作用。在低温T=10~17 K和T=7 K下,交流磁化率测量显示配合物41虚部交流磁化率出现了频率依赖性(图 10中),并且随频率有峰位置的移动,通过拟合得到其有效能垒为53.5 K,弛豫时间为2.3×10-8 s[47];并且出现了低温磁滞回线(图 10右),但矫顽场较低。
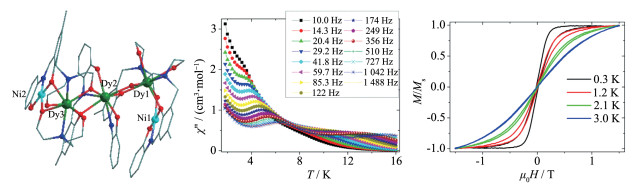
2014年,Long等报道了一系列[Ln2Ni2]四核异金属簇配合物Ln2Ni2L10(bipy)2(Ln=La (45)、Ga (46)、Tb (47)、Dy (48)、Ho (49)),单晶X射线结构分析表明5个配合物为同构型的。配合物48(图 11左)属于单斜P21/c空间群,其不对称单元由一个独立的NiⅡ阳离子,一个独立的DyⅢ阳离子,一个2,2′-联吡啶分子和5个3,5-二氯苯甲酸阴离子组成。有意思的是,4个金属离子之间均通过羧基桥联成线型结构,其中2个DyⅢ之间通过4个双齿配位模式的2,6-二氯苯甲酸(L-)桥联,而DyⅢ与NiⅡ之间通过2个双齿配位模式的L-和一个三齿配位模式L-上的一个羧基O桥联。配合物48中每个NiⅡ离子与3个L-阴离子中的4个氧原子和一个2,2′-联吡啶中的2个N原子进行配位,形成了一个扭曲的NiO4N2八面体配位环境;DyⅢ离子与7个L-阴离子中的7个氧原子配位组成了单帽三棱柱构型,其中O6#1、O8、O9在一个三角平面上;O7#1、O5和O2在另一个三角平面上;O4在帽的位置。交流磁化率研究表明,配合物48不管是在温度还是频率上,都出现了显著地频率依赖现象(图 11右),其有效能垒为105 K[48]。据我们所知,该配合物能垒在Ni-Dy单分子磁体中是最高的。
2017年田金磊等报道了2个Ni - Dy异金属配合物[(μ3-CO3)2{Ni(HL)(CH3CH2OH)Dy(CH3COO)}2]·2CH3CH2OH (49)和[Ni(HL) (H2O) (tfa)Dy(hfac)2] (50,H3L=N,N′-bis(3-methoxysalicylidene)-1,3-diamino-2-prop-anol,Hhfac=六氟乙酰丙酮,tfa-=三氟乙酸阴离子)。配合物49的结构可看成由2个[Ni(HL) (CH3CH2OH)Dy(CH3COO)]2+双核片段通过2个羧基桥联起来。在每个双核片段中,NiⅡ和DyⅢ离子通过2个酚羟基O原子桥联,其中NiⅡ离子采取六配位八面体配位环境,而DyⅢ采取DyO8三角十二面体配位环境。配合物50为中性配合物[Ni(HL)(H2O)(tfa)Dy (hfac)2],其中NiⅡ和DyⅢ之间通过HL2-配体上酚羟基O和一个三氟乙酸阴离子的2个O桥联。NiⅡ同样采取六配位八面体构型,而DyⅢ与49不同,采取九配位“玛芬” (muffin)构型。配合物49在0 Oe外场下均没有明显的频率依赖。配合物49在1 200 Oe dc场下,在311~2 311 Hz出现频率依赖现象,有单分子磁体行为,Ueff=11.52 K。配合物50在0 Oe外加场时有微弱频率依赖,但没有峰值;当加2 000 Oe外加场时,只有在频率大于811 Hz才出现交流磁化率虚部的峰值,排除了自旋玻璃行为,Ueff=5.14 K。配合物49和50弛豫行为不同,可能源于其中DyⅢ配位几何对称性及配位场强度的不同,也为较少的Ni-Dy单分子磁体添加了新的成员[49]。
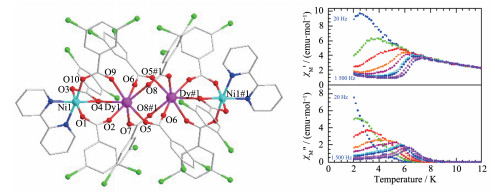
2018年陈自卢等基于1,3-二氨基-2-丙醇配体合成了一例Ni-Dy具有单分子磁体行为的配合物[Ni4Dy2(μ3-OH)2L4(OAc)8]·H2O (51)。它可看成为S型双口袋骨架结构(图 12左下),2个DyⅢ离子被一个“椅”式形状的Ni4O4单元隔开。所有NiⅡ离子采取NiO4N2八面体配位,而DyⅢ采取DyO8反四棱柱构型。配合物51内2个NiⅡ和一个DyⅢ由一个L-配体中的μ3-O桥联在一起,且L-上的2个氨基N原子同时配位2个NiⅡ离子(图 12上a);μ3-OH-也与2个NiⅡ和一个DyⅢ配位(图 12上b)。更有意思的是,醋酸根离子有3种配位模式(图 12上c~e)。这些丰富的配位模式为该新颖结构的形成起到关键作用。把DyⅢ离子换成抗磁性YⅢ离子,得到该配合物的类似物[Ni4Y2]。对[Ni4Y2]类似物磁性拟合表明,Ni1与Ni2及Ni1与Ni2A之间为反铁磁性耦合,而Ni2和Ni2A之间为铁磁性耦合。交流磁化率研究表明,配合物51有频率依赖现象(图 12右下),Cole-Cole图几乎为半圆形,且具有SMM行为,Ueff=11.2 K[50]。
CuⅡ离子采取3d9电子构型,在一般八面体配位场中电子排布为t2g6eg3,尽管CuⅡ的eg上的2个轨道排布3个电子会导致CuⅡ发生Jahn-Teller效应,但对于CuⅡ来说,其磁各向异性并不是很强,甚至一般看成各向同性离子。此外,CuⅡ只有一个未成对电子,自旋为1/2。理论上,该离子不会引起人们在设计3d-4f单分子磁体时的注意。然而,令人惊奇的是,合成Cu-Ln单分子磁体一直受到人们的重视,主要基于以下几点:(1)如前所述,第一个3d-4f单分子磁体为Cu-Tb双核异金属配合物,吸引了人们合成其他核数Cu-Ln单分子磁体;(2)大量研究表明,CuⅡ与GdⅢ在酚羟基O桥联下一般表现出铁磁性耦合,人们期望合成的Cu-Dy配合物中CuⅡ与DyⅢ之间也为铁磁性耦合并提高Cu-Dy单分子磁体性能,因为铁磁耦合有利于抑制量子隧穿效应和提高能垒;(3)合成了50~60个具有单分子磁体性质的Cu-Dy配合物,其数目相对较多,且结构丰富;(4)由于CuⅡ各向同性特征,有利于设计Cu-Gd磁致冷材料,并在最近几年引起重视:在设计Cu-Gd磁致冷材料时,合成Cu-Dy类似物并研究其单分子磁体性能。
2019年Ghosh等报道了4个双核Cu-Ln异金属配合物[(CuL)Ln(NO3)3](H2L=N-α-methylsalicylidene-N′ - 3 - methoxysalicylidene - 1,3 - propanediamine,Ln= Tb (52)、Dy (53)、Ho (54)和Er (55))。这4个配合物结构类似,L配体上2个N原子和2个酚羟基O原子配位到CuⅡ上,形成CuL片段;然后此CuL片段再通过配体L上的2个酚羟基O和配体L上一个甲氧基O原子桥联到LnⅢ上,且LnⅢ的其他配位点被3个NO3- (配位模式为κ2O,O′)的6个O占据。因此,Cu采取CuN2O2平面四方形构型,而LnⅢ采取LnO9畸变的三帽三棱柱空间构型。通过配合物53中Cu…Dy之间键参数和报道的相关文献比较,作者认为Cu…Dy之间为铁磁性耦合。在无外加场时,53没有显示明显的频率依赖信号,然而,加2 000 Oe外加场时,可观测到有单分子磁体行为的频率依赖信号,有效能垒Ueff=53(2) K,其数值比52的Ueff=28.5(5) K高[51]。此外,据我们所知,该文中配合物53的能垒也比绝大多数Cu-Dy单分子磁体能垒高,这或许与53中DyⅢ采取较高对称性的三帽三棱柱空间配位构型有关。
2017年我们使用2,2-二吡啶酮与三乙醇胺作为混合配体合成了一系列[Cu6Ln2]配合物[52][Cu6Ln2 (tea)2{(py)2CO2}3Cl6] ·xH2O(Ln=Dy (56)、Tb (57)、Ho (58)、Er (59)、Gd (60);x=1 (56、58、60)、0 (57)、x=7 (59);(py)2CO2H2=2,2-二吡啶酮二水合物,teaH3=三乙醇胺)。它们结构类似,以配合物56为例,Dy1、Cu1、Cu1d和Cu1e形成一个压缩四面体,3个Cu处在压缩四面体的正三角形底边上;而Dy1a,Cu1a,Cu1b和Cu1c形成另一个类似的压缩多面体;且2个Cu3正三角形底边几乎平行。6个CuⅡ均采用CuNO2Cl四配位平面四方形空间构型,而2个DyⅢ采取少见的DyNO6七配位单帽八面体配位空间构型,但畸变严重。Cu-Gd (60)磁性拟合表明,CuⅡ与GdⅢ离子之间(J1)及2个GdⅢ离子之间(J3)为铁磁性耦合,而CuⅡ离子之间(J2)为反铁磁性耦合。这5个配合物在0 Oe和外加场下,均没有频率依赖现象。为了调控弛豫行为,我们使用席夫碱配体H2L(H2L=N1, N3-bis(3- methoxysalicylidene)diethylenetriamine)替换2,2-二吡啶酮,得到一系列新的[Cu3Ln2]配合物[Cu3Ln2(L)2(teaH)2(N3)2Cl2]·xCH3CN(x=3,Ln=Ho (61)和Gd (62);x=2,Ln=Dy (63))[53]。该同系物5个金属原子形成“风筝”型拓扑结构,2个LnⅢ和一个CuⅡ在“风筝”身体部位,而另2个CuⅡ处在“风筝”翼上(图 13左)。Cu1采取CuN2O2四配位平面正方形空间构型;而Cu2和Cu3采取CuO2N2Cl五配位四角锥构型,其中Cl-处在轴向位置。2个DyⅢ均为DyN2O6八配位,Dy1和Dy2分别采取畸变的双帽三棱柱和三角十二面体构型。Cu-Gd (62)磁性拟合表明,CuⅡ离子之间(J3)和GdⅢ离子之间(J1)为反铁磁性耦合,而CuⅡ与GdⅢ之间(J2)为铁磁性耦合。0 Oe外加场和2 000 Oe外加场对[Cu3Dy2] (63)影响不大,均表现频率依赖但没有峰值,能垒只有4.9 K。这2个系列配合物单分子磁体性能较弱主要源于它们的DyⅢ配位几何对称性较低。
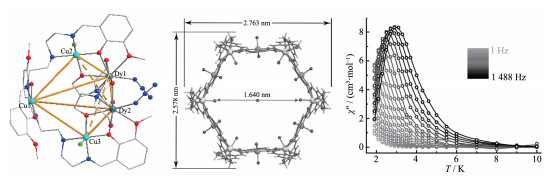
2016年唐金魁课题组报道了一个[Dy6Cu6]环状配合物[Ln6Cu6(H2L′)6Cl12(H2O)6](ClO4)(OH)·30H2O(Tb (64)和Dy (65))。如图 13中所示,配合物64中含有6个(H2L′)2-配体、6个TbⅢ离子、6个CuⅡ离子、6个配位水分子和12个Cl-离子。6个TbⅢ离子排布在六边形的6个顶点处,起到连接相邻的2个六角形边的作用;6个CuⅡ离子装在(H2L′)2-配体的N-N-N配位“口袋”处,且6个CuⅡ离子处在六边形的6条边的中心。有意思的是,该六边形的直径可达到1.640 nm。此外,6个TbⅢ均采用单帽反四棱柱空间配位构型。交流磁化率研究表明,配合物65有单分子磁体行为(图 13右),Ueff=19.6 K[54]。
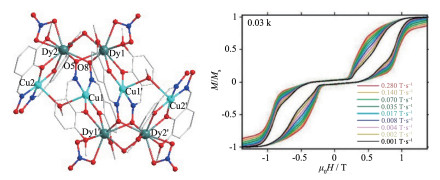
2014年,Powell等报道了一个[CuⅡ4DyⅢ4]的配合物[CuⅡ4DyⅢ4(vanox)6(Hvanox)2(NO3)4(μ-HOMe)2] ·6MeOH(66,H2vanox=3 - methoxy - 2 - hydroxybenzaldoxime) [55]。如图 14左所示,该配合物可看成由4个[CuDy(vanox) (Hvanox)]2+单元组成。在每个单元中,CuⅡ离子与2个肟基N原子和2个酚羟基O原子配位,而DyⅢ离子处在更大的配位“口袋”处,即由2个酚羟基O原子和2个甲氧基O原子配位。[CuDy(vanox)(Hvanox)]2+单元再通过肟基O原子桥联2个相邻的[CuDy (vanox)(Hvanox)]2+单元(如O5和O8可把[Cu1Dy1′]单元和相邻的[Cu2Dy2]和[Cu1′Dy1]连接起来)。交流磁化率研究表明,χ′在高频时有2个峰,表明配合物66存在分子的和配合物内单离子的2种弛豫机制。使用Arrehenius公式拟合高温区τ对温度的变化曲线,得到Ueff=41.6 K。由于无法单独拟合每个弛豫机制,因此该能垒实际为2个弛豫机制相应能垒的平均值。此外,在0.03 K时可观察到该配合物有明显的磁滞回线(图 14右),但该磁滞回线新颖之处在于它出现2个交叉点。如图 14右所示,第一个交叉点(即不同扫场速度时不同的磁滞回线重合)出现在低场处(约0.3 T),表明分子基态为非磁性的。磁性拟合发现[CuDy(vanox)(Hvanox)]2+单元内CuⅡ与DyⅡ之间为铁磁性,而不同[CuDy(vanox)(Hvanox)]2+单元的CuⅡ之间及CuⅡ与DyⅢ之间为反铁磁性耦合,因此,低磁场时,每个离子磁矩抵消而使分子表现非磁性。第二个交叉点出现在高场(约0.7 T)处,这主要由于高场克服了反铁磁性耦合使整个分子处在铁磁态造成的。
如前所述,使用顺磁性3d离子与Dy构建单分子磁体时,配合物内顺磁性3d离子之间及其与Dy之间会存在弱的磁耦合作用,导致量子隧穿效应增大,从而降低单分子磁体有效能垒和阻塞温度。为了解决这一问题,人们尝试使用第一过渡系抗磁性离子CoⅢ或ZnⅡ与Dy构建单分子磁体,以消除3d-4f配合物内3d离子间及3d和Dy之间的磁耦合作用。如果配合物内DyⅢ离子之间通过抗磁性3d隔开,则配合物会表现出更高能垒的单离子磁体性质。人们把这种掺杂抗磁性离子的方法称为磁稀释,其作用与人们在研究有磁耦合的多核Dy配合物时,使用90%或95%的Y替代Dy来提高相应配合物能垒的作用类似。因此,最近五年来,使用第一过渡系抗磁性离子与Dy构建单分子磁体引起人们越来越多的重视,报道的相应配合物的数量急剧增加。人们选择的第一过渡系抗磁性离子一般为CoⅢ和ZnⅡ,因此,在该部分我们分别综述CoⅢ-Dy和ZnⅡ-Dy单分子磁体最新研究情况。
CoⅢ-Dy单分子磁体在近些年研究较多,其中CoⅢ离子是抗磁性离子。因此,CoⅢ-Dy配合物表现的磁弛豫行为可与纯稀土单分子磁体作比较。此外,如CoⅢ-Dy配合物内DyⅢ之间被一个或数个CoⅢ隔开,配合物磁性质可看成是配合物内独立的单个DyⅢ表现出来的,配合物表现出的磁弛豫行为可看成单离子磁体。
文献调研发现,几乎第一过渡系所有离子(包括+2和+3价)之间或其与4f离子之间均可形成蝴蝶型或双缺口立方烷型[M2M′2]结构配合物,其中有些配合物具有优异的单分子磁体性能。有意思的是,在抗磁性CoⅢ与DyⅢ形成的众多拓扑结构单分子磁体中,“蝴蝶”型[CoⅢ2DyⅢ2] SMMs也是研究最为详细的一类,主要涉及调控配体以改变CoⅢ或DyⅢ配位环境、CoⅢ离子及CoⅢ被其他抗磁性离子取代后对整个配合物磁弛豫行为影响等,所得到的这类配合物能垒可从14 K调控到170 K。如2017年Murray等报道了一个[CoⅢ2DyⅢ2]配合物[56][CoⅢ2DyⅢ2(μ3-OH)2(o-tol)4(mdea)2(NO3)2] (67,o-tol-=邻甲基苯甲酸阴离子,mdeaH2=N-甲基二乙醇胺)。配合物内2个CoⅢ和2个DyⅢ通过2个μ3-OH-桥联形成“蝴蝶”型拓扑结构,其中DyⅢ占据“蝴蝶身体”位置,CoⅢ占据“蝴蝶翼”的位置。CoⅢ采取六配位扭曲八面体构型,而DyⅢ采取八配位扭曲反四棱柱构型。交流磁化率研究表明,配合物67有单分子磁体性质,使用多弛豫过程(包括量子隧穿过程、直接过程、Raman过程和Orbach过程)拟合,得到Ueff=116.9(2) K。量子化学计算研究发现,把配合物内抗磁性CoⅢ去掉,配合物基态横向磁各向异性增大,进而导致QTM增大从而降低SMM性能。这表明,尽管CoⅢ为抗磁性,但它对于能否观察到配合物整体的磁弛豫行为至关重要。此外,该论文还通过理论研究模拟了配合物内抗磁性CoⅢ被其他抗磁性离子如ZnⅡ、KⅠ取代对弛豫行为的影响,发现使用ZnⅡ、KⅠ取代CoⅢ后会使SMM性能更好,原因在于这些取代的离子有更强的静电电荷极化作用。
2015年,梁福沛等使用2-(2,3-dihydroxpropyl- iminomethyl)-6-methoxyphenol(H3L)与新特戊酸(Hpiv)得到一个新颖结构的[CoⅢ2DyⅢ4]配合物[CoⅢ2DyⅢ4 (HL)2(μ3-OH)2(piv)10(OH2)2]·2Hpiv·2CH3OH·5H2O (68)。如图 15左所示,配合物内2个CoⅢ与其中2个DyⅢ通过2个μ3-OH-连接同样形成一个“蝴蝶”型[CoⅢ2DyⅢ2]单元,然后该[CoⅢ2DyⅢ2]单元通过HL2-配体上的羟基氧原子和piv-上的羧基O原子桥联另外2个DyⅢ离子。配合物内2个CoⅢ采取六配位扭曲八面体构型,Dy1与Dy1A采取八配位扭曲反四棱柱构型,而Dy2和Dy2A采取少见的七配位单帽三棱柱构型。交流磁化率研究表明配合物68能垒只有18.4 cm-1。从头计算表明其中一个DyⅢ的基态与第一激发态能级尽管达到305.2 cm-1,但其实际能垒只有18.4 cm-1,可能原因在于其中2个DyⅢ之间有弱的磁耦合作用[57]。
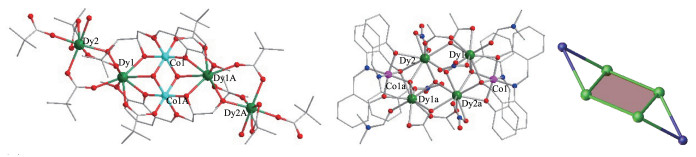
2016年李亚红等使用2-((2-(hydroxymethyl)phe- nylimino)methyl)phenol (H2L)合成了一个配合物[DyⅢ4CoⅢ2(μ3-OH)2L4(NO3)4(CH3COO)4(DMF)2] ·2C2H5OH (69)。有意思的是,配合物69内4个DyⅢ离子与2个μ3-OH-同样可形成一个“蝴蝶”型(或双缺口立方烷)单元,然后该单元通过2个醇O原子和2个酚O原子连接到2个CoⅢ上(图 15中、右)。2个CoⅢ也为六配位扭曲八面体构型,而4个DyⅢ采取单帽反四棱柱构型。交流磁化率研究表明配合物69有效能垒为41.9 K,这在当时已报道的[Dy4] SMMs中算是较高的[58]。
2019年我们使用N1,N3-bis(3-methoxysalicyli- dene)diethylenetriamine(H2L)和2,6 -吡啶二甲醇(pdmH2)混合配体获得一个[CoⅢ2DyⅢ2]配合物[Co2Dy2 (L)2(pdm)2(CH3COO)2(CH3OH)2] (NO3)2·5CH3OH·2.5H2O (70) [59]。如图 16左所示,配合物内2个CoⅢ与2个DyⅢ通过L2-酚基O和甲氧基O及pdm2-上的醇基O桥联形成一个新颖的线型[CoⅢ2DyⅢ2]拓扑结构,其Co1-Dy1-Dy1a的夹角为141.82(2)°。2个CoⅢ同样采取六配位扭曲八面体构型,而2个DyⅢ采取九配位“玛芬”构型。交流磁化率研究表明,该配合物70的Ueff为64.0(9) K。此外,我们也把这个线型[CoⅢ2DyⅢ2]配合物与“蝴蝶”型[CoⅢ2DyⅢ2]配合物进行结构与磁性比较,发现我们报道的线型配合物的Dy-O-Dy键角和Dy-Dy距离均比“蝴蝶”型配合物小,这意味着“蝴蝶”型配合物DyⅢ之间磁耦合作用可能更小,它们表现得更具有单离子磁体的行为。因此,配合物70的能垒会比一些“蝴蝶”型[CoⅢ2DyⅢ2]低。
除“蝴蝶”型[CoⅢ2DyⅢ2]配合物外,还有一些其他拓扑结构配合物。如2012年唐金魁等报道一例以[DyⅢ(HBpz3)2]2+为底物加入过渡金属Co而得到的配合物[CoⅢDy3Ⅲ(HBpz3)6(dto)3] ·4CH3CN·2CH3Cl2 (71),其结构为螺旋桨形,如图 16中所示。配合物内单个CoⅢ离子处于“螺旋桨”中心,而3个DyⅢ离子处于“螺旋桨”的翼的位置。CoⅢ为六配位八面体构型,DyⅢ为八配位反四棱柱构型。配合物71的Ueff为52 K,远大于其FeⅢ类似物的有效能垒15 K,可能与抗磁性CoⅢ与周围3个DyⅢ无磁耦合作用,而低自旋FeⅢ(S=1/2)与DyⅢ之间有弱的磁耦合作用有关[60]。
2016年,Murray等也报道了2个其他拓扑结构配合物[CoⅢ4DyⅢ4(μ-F)4(μ3-OH)4(o-tol)8(mdea)4]·3H2O·EtOH·MeOH (72)和[CoⅢ4DyⅢ4(μ-OH)4(μ3-OMe)4(O2CC (CH3)3)4(tea)4(H2O)4]·4H2O (73)[61]。这2个结构与前述的[CrⅢ4DyⅢ4]配合物有类似之处。此外,这2个结构尽管配位的有机配体不同,但核结构类似,区别在于72使用4个μ-F-桥联相邻2个DyⅢ离子,而73采用4个μ-OH-桥联相邻2个DyⅢ离子。2个配合物均为4个DyⅢ通过OH-和/或F-桥联形成一个Dy4平面四边形单元,然后通过有机配体把此Dy4四边形上的每个DyⅢ连接一个CoⅢ离子(图 16右)。配合物72和73的CoⅢ同样为六配位扭曲八面体,所有DyⅢ均采取八配位双帽三棱柱空间构型。配合物72具有单分子磁体行为,其Ueff为39 K,而配合物73在2 K以上没有频率依赖。理论计算表明,用μ-F-取代μ-OH-可淬灭基态量子隧穿效应。因此,72的SMM行为比73好。此外,进一步理论研究发现,尽管72中含μ-F-使得其基态QTM淬灭,但还存在激发态的QTM,当用顺磁性CrⅢ时,CrⅢ-DyⅢ之间磁耦合作用可进一步降低QTM,这也是72的能垒没有相应的Cr的类似物高的原因。
ZnⅡ离子的d轨道上有10个电子,是抗磁性离子,导致Zn-Dy之间没有磁耦合作用。因此,该领域本不会引起大家太多的注意。然而,截至目前,已经报道了大约100个左右的Zn-Dy单分子磁体配合物,且报道的Zn-Dy SMM配合物数目的还在以惊人的数目增长。该研究领域受到较高重视可能源于以下几个原因:(1)与相对应的M-Ln(M=Co、Ni)配合物和多核Dy配合物相比较而言,Zn-Ln配合物具有较高的Ueff值,可能由于抗磁性ZnⅡ取代了M-Ln位点中顺磁性MⅡ(如CoⅡ或NiⅡ)或多核Dy配合物中某些DyⅢ位点后,使得他们之间的磁耦合消失,从而起到对剩余DyⅢ的磁稀释作用;(2) ZnⅡ与DyⅢ离子可形成拓扑结构多样的Zn-Dy单分子磁体配合物;(3)通过配位(或晶格)溶剂分子或离子的替换或脱除调控Dy或Zn的配位几何,进而调控配合物磁弛豫性能。当然,还有一些课题组研究了配合物内ZnⅡ数目对Zn-Dy配合物弛豫行为的影响等。
2013年童明良等报道了一个[Zn2Dy]配合物,其结构式为[Zn2DyL2(MeOH)]NO3·3MeOH·H2O (74,L=2,2′,2″-(((nitrilotris(ethane-2,1-diyl))tris(azanediyl)) tris(methylene))tris - (4 - bromophenol))。如图 17上所示,通过配体L的配位O或N原子把DyⅢ夹在2个ZnⅡ之间。有意思的是,如将74放置在空气中干燥1 d,使其失去配位的甲醇分子,则得到配合物[Zn2DyL2]NO3·H2O (75);反之,如把75放在甲醇溶液中1 d,也可以得到配合物74。如图 17上所示,配合物75与74的区别在于75的DyⅢ配位点上无甲醇配位分子。通过SHAPE软件计算发现,DyⅢ离子的配位几何构型由配合物74的五角双锥(quasi-D5h)转变为配合物75的八面体(quasi-Oh)。如图 17下所示,配合物74在0 Oe外加场下便有SMM性质,而75只有在1 200 Oe外加场下才有该性质,且配合物74的Ueff数值439 K比75的有效能垒64 K高7倍。此外,对理想的五角双锥和理想的正八面体配位几何片段(分别为[Dy(OCH)7]4-和[Dy(OCH)6]3-)分别进行从头计算发现,五角双锥配位几何具有完美的轴向对称性(gx=gy=0),这使得这种配位几何能完全抑制量子隧穿效应,并能使相应配合物成为性能优异的单离子磁体。然而,正八面体配位几何呈现各向同性特征(gx=gy=gz=6.58),使得相应配合物具有极快的磁弛豫行为。对于配合物75,DyⅢ配位几何偏离完美八面体而使之具有一定的轴向对称性,因此,该配合物也会表现出一定的慢弛豫行为[62]。
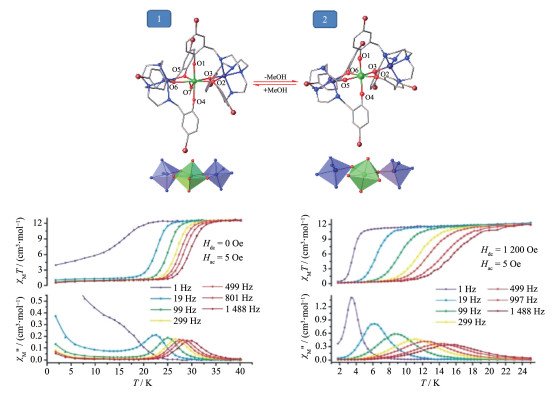
2016年,孙文彬等使用Zn基前驱体[ZnL1]分别与DyCl3·6H2O或DyBr3在甲醇或乙醇溶液中加热回流分别得到3个Zn-Dy配合物,即[Zn2(L1)2DyCl3]·2H2O (76)、[Zn2(L1)2Dy(MeOH)Br3]·3H2O (77)和[Zn2(L1)2Dy (H2O)Br2][ZnBr4]0.5 (78),并用[ZnL2]与DyCl3·6H2O在甲醇中反应得到另一个Zn - Dy配合物[Zn2(L2)2DyCl3]·2H2O (79)(配合物76~79的合成路线及其中的H2L1或H2L2配体如图 18左所示)[63]。配合物76~ 79中的ZnⅡ与处在(L1)2-或(L2)2-配体“内侧”的N2、O2原子配位,另有Cl-或Br-配位在ZnⅡ的轴向位置,因此,4个配合物中的ZnⅡ均采取五配位四角锥构型。而配合物76~79中的DyⅢ与2个[ZnL1]或[ZnL2]片段上的8个O配位,每个片段上4个O原子均为(L1)2-或(L2)2-“外侧”的O2、O2原子。此外,每个配合物中DyⅢ的第9个配位点被Cl-、H2O或CH3OH占据。有意思的是,配合物中4个甲氧基上的O和1个配位的分子或离子几乎在DyⅢ周围形成一个五元环。76~ 79中的DyⅢ严重偏离理想的单帽反四棱柱、三帽三棱柱及“玛芬”构型(CShM在2.1~3.6之间),且偏离程度相似。磁性研究表明,配合物76~79的能垒分别为481、233、121和398 K。从头计算表明77和78的gx和gy比另2个配合物的大,说明77和78的横向晶体场参数比76和79大,导致77和78的量子隧穿效应较大,因此它们的能垒比76和79的低。更有意思的是,4个配合物均有磁滞回线,阻塞温度分别为8、6、4和8 K(图 18右)。
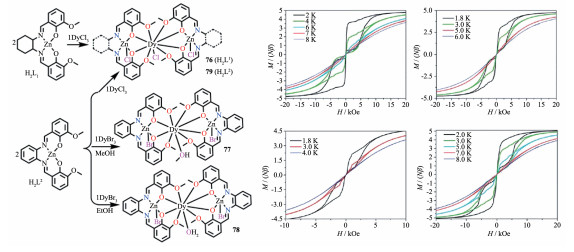
2015年,Colacio等使用席夫碱配体N,N′ - dimethyl-N, N′-bis(2-hydroxy-3-formyl-5-bromo-benzyl) - ethylenediamine(H2L),得到了[Zn2Dy]三核配合物[ZnCl(μ-L)Dy(μ-L)ClZn]PF6 (80)[64]。配合物内的1个DyⅢ通过2个μ-L2-配体与处于DyⅢ两侧的2个ZnⅡ连接起来,形成“V”型结构。有趣的是,该“V”型结构的开口处正好“钳”住一个PF6-阴离子。此外,该配合物还有一个正好通过Dy和P的C2轴。配合物80中ZnⅡ采取五配位四角锥构型,而Dy采取DyO8八配位,其配位几何介于反四棱柱、三角十二面体和双帽三棱柱理想几何构型之间。在0 Oe外加场时,其能垒达到186 cm-1,当加1 000 Oe外加场时,其能垒可提高到222 cm-1。此外,配合物80的能垒几乎是另一个当时已报道的[ZnCl(μ-L)Dy(μ-L)ClZn][ZnCl3 (CH3OH)] (81)的能垒的2倍。通过结构比较发现,配合物80和81的区别仅在于80的阴离子为PF6-,而81为[ZnCl3(CH3OH)]-。理论计算表明,对于配合物80,[ZnCl(μ-L)Dy(μ-L)ClZn]+对称性更高,有通过DyⅢ的C2轴,这使得其2个最低能量的KD态(Kramers doublet)的各向异性轴几乎共线,从而使得配合物80的第一激发态的KD态被抑制,导致热弛豫过程只能通过第二激发态来实现。因此,配合物80的Ueff接近于配合物81的Ueff的2倍。该研究表明,通过更换不同对称性和/或体积大小的阴离子可实现调控配合物有效能垒的目的。
同年,Colacio等又使用席夫碱配体H2L(H2L=N,N′-2, 2-dimethylpropylenedi(3-methoxysalicylideneimi- nato))合成了3个[Zn2Dy]配合物:[(LZn(H2O))2Dy(H2-O)](CF3SO3)3 (82)、[(LZnBr)2Dy(H2O)]ClO4 (83)和[(LZn-Cl)2Dy(H2O)]ClO4·MeOH (84)。82~84的区别在于其上的ZnⅢ分别被配位水、Br-和Cl-配位,这导致它们的阴离子也不相同。82~84均使用L2-配体上“内侧”的2个N原子和2个酚羟基O原子螯合一个ZnⅡ离子占据ZnⅡ赤道面,形成一个ZnL单元;然后2个ZnL单元分别用它们L配体“外侧”的2个酚羟基O和2个甲氧基O原子与处于2个单元之间的DyⅢ配位。最后,每个ZnⅡ的轴向位置被H2O、Br-或Cl-占据,而DyⅢ的一个配位点也被配位水占据。因此,3个配合物的每个ZnⅡ均采用五配位四角锥构型。配合物82~84中DyⅢ虽然都是九配位,但82中DyⅢ为“玛芬”构型,而83和84则采取三帽三棱柱构型。3个配合物均有单分子磁体行为,1 000 Oe外加场时得到的82~84的Ueff分别为128.6(5)、214.7和202.4 K。理论计算表明,3个配合物的较大能垒源于ZnⅡ的存在导致桥联的酚羟基O原子有大的电子极化;此外,配合物内没有磁耦合作用和QTM的淬灭也是导致它们能垒较高的原因[65]。
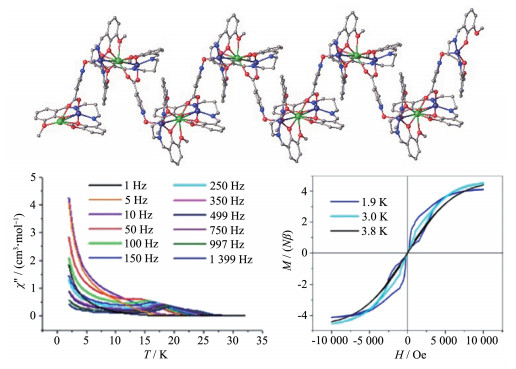
此外,如果使用适当的桥联配体能把[Zn2Dy]配合物单体桥联成链状结构,研究其弛豫行为,也是一个很有意义的事情,因为含抗磁性ZnⅡ的配合物的SMM性质主要源于DyⅢ弛豫,改变DyⅢ配位环境,有可能对配合物弛豫行为会有很大影响,但要解决2个问题:(1)单体要有合适的容易取代的配位点。我们知道,很多[Zn2Dy]配合物都有一些小配位分子或离子如NO3-、H2O、X-(卤素离子),这为通过桥联配体取代小分子提供了便利。(2)找到合适的桥联配体,要求不能对单体上配体有太大空间位阻。实际上,2018年朱道本等选择合适多齿螯合席夫碱配体(H2L=N,N′-bis(3-methoxysalicylidene)-1,3-diamino- propane)便构建了一个[DyZn2(salen)2]单元,然后这些单元通过桥联配体POC-(POC-=pyridin-N-oxide-4-carboxylate)成功地形成了一个“Z”型一维链{[Zn2Dy (L)2(POC)](OH)(ClO4)}·H2O·MeOH (85,图 19左)。该一维链在0 Oe外场下,就有SMM行为(图 19中),其Ueff为235.3 K;而在1 000 Oe直流场下,其Ueff进一步提高,达到372.4(9) K。此外,该配合物在3.8 K时有“蝴蝶”型的磁滞回线[66](图 19右)。由于该一维链单体之间距离较远,性质可看成SMM或SIM,但不能看成单链磁体。然而,需要指出的是,该一维链配合物的Ueff在Zn-Dy单分子磁体中是比较高的,同时也说明可通过桥联配体进一步调控单体弛豫行为。
除了上述的[Zn2Dy]配合物或以Zn2Dy为单元构筑的链状配合物外,研究者们还报道了其他核数的配合物,但它们的能垒一般比上述的Zn-Dy配合物低。例如,2016年,Sorace等使用2-甲酰基-6-羟甲基-对甲基苯酚与1,3-二氨基-2-丙醇缩合得到一个席夫碱配体(LH5),然后此配体与Zn和Dy盐反应得到一个双核Zn-Dy配合物,[ZnDy(LH4)2](NO3)3·6H2O(86)[67]。如图 20左所示,一个ZnⅡ和一个DyⅢ通过2个LH4-上的酚O原子桥联(每个LH4-提供一个酚O原子),ZnⅡ和DyⅢ的其他配位点被LH4-上的其他O或N原子配位。ZnⅡ采取六配位八面体构型,而DyⅢ采取反四棱柱构型,CShM计算值为0.619。这在多核配合物中还是比较小的,这可能是配合物86的能垒较高的原因之一。配合物86在零场和外加直流场均显示SMM行为,Ueff为189 cm-1,是目前双核[ZnDy]中配合物中能垒最高的。此外,在2 K下,可观察到配合物86的“蝴蝶”型磁滞回线。作者还研究了使用Y进行磁稀释对配合物弛豫行为的影响。但研究发现,即使把DyⅢ稀释到1%,在零场下配合物弛豫行为也没有明显变化,可能由于晶格内DyⅢ距离过短,导致尽管进行了磁稀释,晶格内DyⅢ之间的距离还是较短。
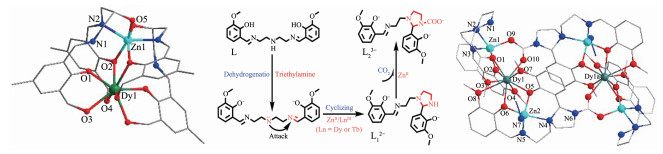
2019年,我们使用席夫碱配体N1,N3 - bis(3- methoxysalicylidene)diethylenetriamine(L)合成了一个具有单分子磁体行为的[Zn4Dy2]配合物[Zn4Dy2(L1)2(L2)2(N3)2]Cl2·2H2O (87)。如图 20中所示,在配合物87形成过程中,L配体首先关环形成配体L12-,然后L12-原位吸收空气中CO2形成氨基甲酸酯类配体L23-。据我们所知,这是第一个通过吸收空气中CO2形成氨基甲酸酯配体而后形成3d-4f单分子磁体的例子。如图 20右所示,配合物87可看成是通过L12-、L23-配体上N、O配位原子连接2个ZnⅡ离子和一个DyⅢ,形成一个[Zn2Dy]单元,且DyⅢ处于2个ZnⅡ之间。2个[Zn2Dy]单元再通过L23-的羧基O连接起来。对配合物87的Gd的类似物进行磁性拟合发现,尽管2个单元上的Gd的距离较长(约为0.846 nm),但仍存在较弱的反铁磁耦合作用(-0.005 9 cm-1),并且此作用可通过理论计算证实。配合物内ZnⅡ采取五配位四角锥构型,而DyⅢ采取九配位“玛芬”构型。磁性研究表明,该配合物有单分子磁体行为,Ueff的数值为30.66(5) K。理论计算表明,配合物87主要通过第一激发态进行热弛豫[68]。
总之,本文较详细地综述了绝大多数结构新颖、单分子磁体性能优异的第一过渡系金属-镝混金属单分子磁体(TM-Dy SMMs)。通过该综述,我们发现由于大多数顺磁性3d离子与DyⅢ形成的配合物存在弱的磁耦合作用,导致它们的量子隧穿效应增强,进而使得这类配合物的能垒没能得到大幅度提高,尤其对于Mn-Dy和Fe-Dy及Ni-Dy混金属配合物。因此,最近五年来,使用第一过渡系抗磁性金属离子如CoⅢ和ZnⅡ构建单分子磁体引起了人们的重视,相应配合物数目也急剧增加,并获得有效调控磁弛豫行为的手段,如磁稀释、溶剂取代、配体取代等。
此外,需要指出的是,尽管消除磁耦合作用可通过使用抗磁性CoⅢ或ZnⅡ来实现,且得到的相应配合物的能垒比使用顺磁性3d离子有很大提高,但却远远低于部分单核Dy配合物的能垒。这意味着对于TM-Dy SMMs,除了需要消除磁耦合外,至少还有以下3个方面需要我们重点研究:
(1) 提高TM-Dy SMMs内Dy离子的配位几何的轴向对称性以便抑制量子隧穿效应和提高配合物的Ueff和TB。研究发现,当Dy的配位几何具有高次旋转轴Cn(轴次n>7,包括C∞)、C5h/D5h(如五角双锥或五棱柱)、S8/D4d(如反四棱柱或双帽反四棱柱)及S12/D6d(反六棱柱)时[69],所有横向晶体场参数会消失,进而导致配合物的量子隧穿效应被有效抑制。基于这一理论,目前人们已经报道了多例高性能Dy-单离子磁体[18]。然而,除少数TM-Dy SMMs(如[Fe2Dy]、[Zn2Dy]等)中的Dy的配位几何得到有效控制外,绝大多数这类配合物中的Dy配位几何对称性较低或偏离高轴向配位几何较大,这可能是TM-Dy SMMs的能垒较低的最重要原因。当然,国内外许多课题组或许已经注意到这一问题,但绝大多数TM-Dy配合物的能垒提高效果不理想,这说明控制这类配合物中Dy的配位几何具有很大的挑战性。笔者认为,我们或许可使用一种配位点较多且具有一定配位空间的配体占据TM-Dy配合物中Dy的赤道面位置,然后使用另外一种配位点较少但有一定空间位阻的配体配位Dy的轴向位置来实现配位几何的控制。
(2) 调控TM-Dy SMMs中DyⅢ的基态mJ周围的电荷分布。目前调控TM-Dy SMMs的常用手段有使用有机配体的不同衍生物构筑配合物、配位小分子或离子(如配位H2O、NO3-)或晶格溶剂分子、离子的取代或脱除等。尽管这些方式能对系列配合物的磁弛豫行为有调控作用,并能使系列配合物间有几K至几十K的能垒变化,但还不能对能垒有大幅度调控。除了上述配合物中Dy的配位几何对称性外,Dy周围的配位原子的分布也起到非常重要的作用。按照Rinehart和Long提出的静电作用模型[70],DyⅢ的mJ=15/2的态是扁球形的,而它的mJ=1/2的态是长球形的。为了增强Dy的磁各向异性并使mJ=15/2处于基态,需要降低稀土离子mJ态与配位原子之间的静电排斥。例如,扁球形mJ=15/2的态与处在Dy的轴向(假定z方向)位置的配位原子排斥力小,使mJ= 15/2能量降低而使之处于基态,而长球形的mJ=1/2与DyⅢ的z方向排斥力较大,使mJ=1/2能量升高而使之处于激发态。尽管静电作用模型有一些缺陷,但它对于设计Dy单离子磁体能发挥很好的指导作用,并能较好地控制配合物的能级分布。然而,对于TM-Dy配合物,通过静电模型控制能级分布同样有很大挑战。笔者认为,大多数TM-Dy配合物中DyⅢ与配体原子间排斥力没能很好控制也是导致这类配合物能级较低的原因之一,因为这很可能会导致DyⅢ的态是由不同mJ态混杂而成的,进而增大了量子隧穿效应,从而使得配合物很大可能通过第一激发态弛豫。因此,我们在设计TM-Dy配合物时,至少需要让Dy的赤道面上的配位强度比Dy的轴向的弱,这可能需要使用合适配体或使用2种有机配体组合来实现。
(3)提高TM-Dy SMMs的磁滞回线的面积和矫顽场大小。这2个指标是“磁体”材料需要重点考虑的,可通过提高顺磁离子之间磁耦合作用来实现。众所周知,多核过渡金属配合物中,金属离子之间磁耦合作用大小与金属离子间距离及金属-桥联原子-金属的夹角有关,例如对于多核Cu配合物,当Cu-O-Cu夹角大于97°时,CuⅡ离子之间为反铁磁耦合;而当此夹角小于97°时,CuⅡ离子间为铁磁性耦合,且磁耦合大小也受到CuⅡ间距离及此夹角影响[71]。对于顺磁性3d金属离子与Dy形成的配合物,尽管配合物内3d离子与Dy磁耦合较弱,但研究表明,3d…Dy间磁耦合也与3d…Dy距离及金属离子与桥联原子之间的夹角或二面角有关。例如,Cu… Gd之间磁耦合与Gd-O2-Cu单元中CuO2平面和GdO2平面间的二面角(φ)有关[53]。这意味着需要人们合成更多具有新颖拓扑结构的配合物以调控键参数及磁弛豫行为。此外,笔者认为,使用螯合自由基配体构建配合物使得自由基配位原子桥联在金属原子间,或许也是增强磁耦合的一个重要手段之一。
Supporting information is available at http://www.wjhxxb.cn
Pan Y Z, Hua Q Y, Lin L S, et al. Inorg. Chem. Front., 2020, 7(12):2335-2342 doi: 10.1039/D0QI00326C
Sessoli R, Tsai H L, Schake A R, et al. J. Am. Chem. Soc., 1993, 115(5):1804-1816 doi: 10.1021/ja00058a027
Lin P H, Burchell T J, Clerac R, et al. Angew. Chem. Int. Ed., 2008, 47(46):8848-8851 doi: 10.1002/anie.200802966
Wang H S, Long Q Q, Hu Z B, et al. Dalton Trans., 2019, 48(35):13472-13482 doi: 10.1039/C9DT02638J
王会生, 乐琳, 潘敏.无机化学学报, 2016, 32(1): 153-160 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20160122&flag=1WANG Hui-Sheng, YUE Lin, PAN Min, et al. Chinese J. Inorg. Chem., 2016, 32(1): 153-160 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20160122&flag=1
Wang H S, Song Y. Inorg. Chem. Commun., 2013, 35:86-88 doi: 10.1016/j.inoche.2013.05.016
Murugesu M, Habrych M, Wernsdorfer W, et al. J. Am. Chem. Soc., 2004, 126(15):4766-4767 doi: 10.1021/ja0316824
Stamatatos T C, Abboud K A, Wernsdorfer W, et al. Angew. Chem. Int. Ed., 2007, 46(6):902-906
Tasiopoulos A J, Vinslava A, Wernsdorfer W, et al. Angew. Chem. Int. Ed., 2004, 43(16):2117-2121 doi: 10.1002/anie.200353352
Abbasi P, Quinn K, Alexandropoulos D I, et al. J. Am. Chem. Soc., 2017, 139(44):15644-15647 doi: 10.1021/jacs.7b10130
Stamatatos T C, Abboud K A, Wernsdorfer W, et al. Angew. Chem. Int. Ed., 2006, 45(25):4134-4137 doi: 10.1002/anie.200600691
Ako A M, Hewitt I J, Mereacre V, et al. Angew. Chem. Int. Ed., 2006, 118(30):5048-5051 doi: 10.1002/ange.200601467
Milios C J, Vinslava A, Wernsdorfer W, et al. J. Am. Chem. Soc., 2007, 129(10):2754-2755 doi: 10.1021/ja068961m
Waldmann O. Inorg. Chem., 2007, 46(24):10035-10037 doi: 10.1021/ic701365t
Ruiz E, Cirera J, Cano J, et al. Chem. Commun., 2008(1):52-54 doi: 10.1039/B714715E
Neese F, Pantazis D A. Faraday Discuss., 2011, 148:229-238 doi: 10.1039/C005256F
Kido T, Ikuta Y, Sunatsuki Y, et al. Inorg. Chem., 2003, 42(2):398-408 doi: 10.1021/ic026045d
Guo F S, Day B M, Chen Y C, et al. Science, 2018, 362(6421):1400-1403 doi: 10.1126/science.aav0652
Rinck J, Novitchi G, Van den Heuvel W, et al. Angew. Chem. Int. Ed., 2010, 49(41):7583-7587 doi: 10.1002/anie.201002690
Langley S K, Forsyth C M, Moubaraki B, et al. Dalton Trans., 2015, 44(3):912-915 doi: 10.1039/C4DT03100H
Thuesen C A, Pedersen K S, Schau-Magnussen M, et al. Dalton Trans., 2012, 41(37):11284-11292 doi: 10.1039/c2dt31302b
Langley S K, Wielechowski D P, Vieru V, et al. Angew. Chem. Int. Ed., 2013, 52(46):12014-12019 doi: 10.1002/anie.201306329
Langley S K, Le C, Ungur L, et al. Inorg. Chem., 2015, 54(7):3631-3642 doi: 10.1021/acs.inorgchem.5b00219
Langley S K, Wielechowski D P, Moubaraki B, et al. Chem. Commun., 2016, 52(73):10976-10979 doi: 10.1039/C6CC06152D
Xiang H, Lu W G, Zhang W X, et al. Dalton Trans., 2013, 42(4):867-870 doi: 10.1039/C2DT32651E
Car P E, Favre A, Caneschi A, et al. Dalton Trans., 2015, 44(36):15769-15773 doi: 10.1039/C5DT02459E
Vignesh K R, Soncini A, Langley S K, et al. Nat. Commun., 2017, 8:1023 doi: 10.1038/s41467-017-01102-5
Chen S H, Mereacre V, Zhao Z Y, et al. Dalton Trans., 2018, 47(22):7456-7462 doi: 10.1039/C8DT01289J
Ako A M, Mereacre V, Clerac R, et al. Chem. Commun., 2009(5):544-546 doi: 10.1039/B814614D
Li X L, Min F Y, Wang C, et al. Dalton Trans., 2015, 44(7):3430-3438 doi: 10.1039/C4DT03713H
Lin P H, Tsui E Y, Habib F, et al. Inorg. Chem., 2016, 55(12):6095-6099 doi: 10.1021/acs.inorgchem.6b00630
Wang H S, Yang F J, Long Q Q, et al. Dalton Trans., 2016, 45(45):18221-18228 doi: 10.1039/C6DT02945K
Akhtar M N, Lan Y, Aldamen M A, et al. Dalton Trans., 2018, 47(10):3485-3495 doi: 10.1039/C7DT04304J
Vignesh K R, Langley S K, Moubaraki B, et al. Inorg. Chem., 2018, 57(3):1158-1170 doi: 10.1021/acs.inorgchem.7b02608
Li M Y, Lan Y H, Ako A M, et al. Inorg. Chem., 2010, 49(24):11587-11594 doi: 10.1021/ic101754g
Vignesh K R, Langley S K, Moubaraki B, et al. Chem. Eur. J., 2015, 21(46):16364-16369 doi: 10.1002/chem.201503424
Schmidt S F M, Merkel M P, Kostakis G E, et al. Dalton Trans., 2017, 46(45):15661-15665 doi: 10.1039/C7DT03149A
Tziotzi T G, Kalofolias D A, Tzimopoulos D I, et al. Dalton Trans., 2015, 44(13):6082-6088 doi: 10.1039/C5DT00300H
Liu J L, Wu J Y, Chen Y C, et al. Angew. Chem. Int. Ed., 2014, 53(47):12966-12970 doi: 10.1002/anie.201407799
Chen S H, Mereacre V, Anson C E, et al. Dalton Trans., 2016, 45(22):9336-9344 doi: 10.1039/C6DT01364C
Schmidt S F. M, Koo C, Mereacre V, et al. Inorg. Chem., 2017, 56(9):4796-4806
Han H T, Li X L, Zhu X F, et al. Eur. J. Inorg. Chem., 2017(41):4879-4883
Li J, Wei R M, Pu T C, et al. Inorg. Chem. Front., 2017, 4(1):114-122
Peng Y, Mereacre V, Anson C E, et al. Dalton Trans., 2017, 46(16):5337-5343 doi: 10.1039/C7DT00548B
De Souza M S, Briganti M, Reis S G, et al. Inorg. Chem., 2019, 58(3):1976-1987
Huang G Z, Ruan Z Y, Zheng J Y, et al. Sci. China Chem., 2018, 61(11):1399-1404 doi: 10.1007/s11426-018-9310-y
Chandrasekhar V, Bag P, Kroener W, et al. Inorg. Chem., 2013, 52(22):13078-13086 doi: 10.1021/ic4019025
Zhao F H, Li H, Che Y X, et al. Inorg. Chem., 2014, 53(18):9785-9799 doi: 10.1021/ic501374q
Jiang L, Liu Y, Liu X, et al. Dalton Trans., 2017, 46(37):12558-12573 doi: 10.1039/C7DT02351K
Pei S M, Hu Z B, Chen Z L, et al. Dalton Trans., 2018, 47(6):1801-1807 doi: 10.1039/C7DT04003B
Mahapatra P, Koizumi N, Kanetomo T, et al. New J. Chem., 2019, 43(2):634-643 doi: 10.1039/C8NJ03512A
Wang H S, Yang F J, Long Q Q, et al. New J. Chem., 2017, 41(13):5884-5892 doi: 10.1039/C7NJ00459A
Chen Y, Long Q Q, Hu Z B, et al. New J. Chem., 2019, 43(21):8101-8108 doi: 10.1039/C9NJ00892F
Wu J F, Zhao L, Zhang L, et al. Angew. Chem. Int. Ed., 2016, 55(50):15574-15578 doi: 10.1002/anie.201609539
Kühne I A, Magnani N, Mereacre V, et al. Chem Commun., 2014, 50(15):1882-1885 doi: 10.1039/c3cc46458j
Vignesh K R, Langley S K, Murray K S, et al. Inorg. Chem., 2017, 56(5):2518-2532 doi: 10.1021/acs.inorgchem.6b02720
Zou H H, Sheng L B, Liang F P, et al. Dalton Trans., 2015, 44(42):18544-18552 doi: 10.1039/C5DT03368C
Zhang H F, Liu R, Zhang J, et al. CrystEngComm, 2016, 18(42):8246-8252 doi: 10.1039/C6CE01589A
Wang H S, Yin C L, Hu Z Bo, et al. Dalton Trans., 2019, 48(27):10011-10022 doi: 10.1039/C9DT00774A
Xu G F, Gamez P, Tang J K, et al. Inorg. Chem., 2012, 51(10):5693-5698 doi: 10.1021/ic300126q
Vignesh K R, Langley S K, Murray K S, et al. Chem. Eur. J., 2017, 23(7):1654-1666 doi: 10.1002/chem.201604835
Liu J L, Chen Y C, Zheng Y Z, et al. Chem. Sci., 2013, 4(8):3310-3316 doi: 10.1039/c3sc50843a
Sun W B, Yan P F, Jiang S D, et al. Chem. Sci., 2016, 7(1):684-691 doi: 10.1039/C5SC02986D
Oyarzabal I, Ruiz J, Ruiz E, et al. Chem. Commun., 2015, 51(62):12353-12356 doi: 10.1039/C5CC04495B
Costes J P, Titos-Padilla S, Oyarzabal I, et al. Chem. Eur. J., 2015, 21(44):15785-15796 doi: 10.1002/chem.201501500
Liu C M, Zhang D Q, Su J B, et al. Inorg. Chem., 2018, 57(17):11077-11086 doi: 10.1021/acs.inorgchem.8b01653
Amjad A, Madalan A M, Andruh M, et al. Chem. Eur. J., 2016, 22(36):12849-12858 doi: 10.1002/chem.201601996
Yin C L, Hu Z B, Long Q Q, et al. Dalton Trans., 2019, 48(2):512-522 doi: 10.1039/C8DT03849J
Liu J L, Chen Y C, Tong M L. Chem. Soc. Rev., 2018, 47(7):2431-2453 doi: 10.1039/C7CS00266A
Rinehart J D, Long J R. Chem. Sci., 2011, 2(11):2078-2085 doi: 10.1039/c1sc00513h
Crawford V H, Richardson H W, Wasson J R, et al. Inorg. Chem., 1976, 15(9):2107-2110 doi: 10.1021/ic50163a019

图 1 [Mn12]-OAc的不同mS态的势能图
Figure 1 Plot of potential energy as a function of mS quantum number for [Mn12]-OAc

图 2 通过web of science统计出的2003—2019年期间出版的第一过渡系金属-镝单分子磁体的论文数量
Figure 2 Number of the published papers related to the first series transition metal-Dy single molecule magnets in a range of 2003—2019 through web of science



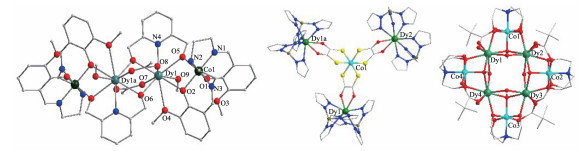
图 5 [MnⅢ12MnⅡ6DyⅢ(μ4-O)8(μ3-Cl)6.5(μ3-N3)1.5(HL)12(MeOH)6] (7, 左)[29]、Dy2Mn(C7H5O2)8(10, 中)[30]和[Mn8Dy2O2(OH)2{(py)2CO2}4(teaH)4(CH3COO)6] (15, 右)[32]的晶体结构
Figure 5 Crystal structures of [MnⅢ12MnⅡ6DyⅢ(μ4-O)8(μ3-Cl)6.5(μ3-N3)1.5(HL)12(MeOH)6] (7, left)[29], Dy2Mn(C7H5O2)8 (10, middle)[30] and [Mn8Dy2O2(OH)2{(py)2CO2}4(teaH)4(CH3COO)6] (15, right)[32]

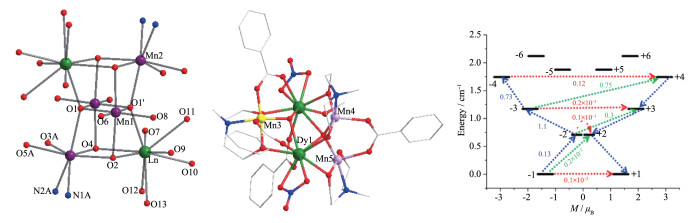
图 6 双立方烷MnⅡ2MnⅢ2LnⅢ2系列配合物核骨架(17, 左)[33]、[MnⅣMnⅢ2DyⅢ2O2(benz)4(mdea)3(NO3)2(MeOH)] (19, 中)晶体结构及其低占据交换谱(右)[34]
Figure 6 Core structure of double-cubane MnⅡ2MnⅢ2LnⅢ2 (17, left)[33], crystal structure of [MnⅣMnⅢ2DyⅢ2O2(benz)4(mdea)3(NO3)2(MeOH)] (19, middle) and its low-lying exchange spectra (right)[34]

图 7 [Fe2Dy(L)2(H2O)]ClO4·2H2O (28, 左)的晶体结构图、χ″ vs ν和χ′ vs ν曲线(中, 插图为阿伦尼乌斯公式拟合图)及其交换基态上金属位点主磁矩方向(右)[39]
Figure 7 Crystal structure of [Fe2Dy(L)2(H2O)]ClO4·2H2O (28, left), plots of χ″ vs ν and χ′ vs ν (middle, inset: plot of relaxation time τ vs T-1 obtained from χM″(ν); Solid lines correspond to an Arrhenius law) and its orientations of the main magnetic moments on metal sites in the ground exchange state (right)[39]


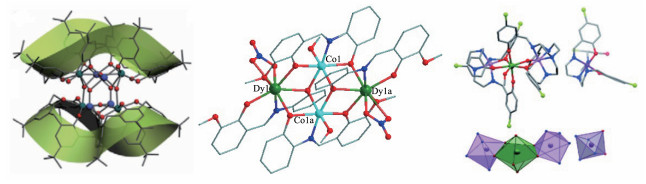
图 9 [(DyⅢ4CoⅡ4)(C8A)2(O)2(DEF)8(H2O)4]·0.5CH3OH (31, 左)[42]、[CoⅡ2Dy2(L)4(NO3)2(MeOH)2]·2CH2Cl2 (35, 中)[44]和[CoⅡ2Dy(TTTTCl)2(MeOH)][CoⅢ(HTTTTCl)](NO3)2·2.5MeOH·2H2O (40, 右)[46]的晶体结构
Figure 9 Crystal structures of [(DyⅢ4CoⅡ4)(C8A)2(O)2(DEF)8(H2O)4]·0.5CH3OH (31, left)[42], [CoⅡ2Dy2(L)4(NO3)2(MeOH)2]·2CH2Cl2 (35, middle)[44] and [CoⅡ2Dy(TTTTCl)2(MeOH)][CoⅢ(HTTTTCl)](NO3)2·2.5MeOH·2H2O (40, right)[46]




图 13 [Cu3Dy2(L)2(teaH)2(N3)2Cl2]·2CH3CN (63, 左)[53]和环状[Tb6Cu6(H2L′)6Cl12(H2O)6] (64, 中)的晶体结构及配合物[Dy6Cu6(H2L′)6Cl12(H2O)6] (65)的χ″ vs T曲线(右)[54]
Figure 13 Crystal structures of [Cu3Dy2(L)2(teaH)2(N3)2Cl2]·2CH3CN (63, left) and ring-like [Tb6Cu6(H2L′)6Cl12(H2O)6] (64, middle) and plots of χ″ vs T for [Dy6Cu6(H2L′)6Cl12(H2O)6] (65, right)[54]


图 15 [CoⅢ2DyⅢ4(HL2)2( μ3-OH)2(piv)10(OH2)2]·2Hpiv·2CH3OH·5H2O的晶体结构 (68, 左)[57], 及配合物[DyⅢ4CoⅢ2( μ3-OH)2L4(NO3)4(CH3COO)4(DMF)2]·2C2H5OH(69)的晶体结构 (中)和核结构 (右)[58]
Figure 15 Crystal structure of [CoⅢ2DyⅢ4(HL2)2( μ3-OH)2(piv)10(OH2)2]·2Hpiv·2CH3OH·5H2O (68, left)[57], crystal structure (middle)and the core (right) of complex [DyⅢ4CoⅢ2( μ3-OH)2L4(NO3)4(CH3COO)4(DMF)2]·2C2H5OH (69)[58]

图 16 [Co2Dy2(L)2(pdm)2(CH3COO)2(CH3OH)2](NO3)2·5CH3OH·2.5H2O (70, 左)[59]、[CoⅢDy3Ⅲ(HBpz3)6(dto)3]·4CH3CN·2CH3Cl2 (71, 中)[60]和[CoⅢ4DyⅢ4(μ-OH)4(μ3-OMe)4(O2CC(CH3)3)4(tea)4(H2O)4]·4H2O (73, 右)[61]的晶体结构
Figure 16 Crystal structures of [Co2Dy2(L)2(pdm)2(CH3COO)2(CH3OH)2](NO3)2·5CH3OH·2.5H2O (70, left)[59], [CoⅢDy3Ⅲ(HBpz3)6(dto)3]·4CH3CN·2CH3Cl2 (71, middle)[60] and [CoⅢ4DyⅢ4(μ-OH)4(μ3-OMe)4(O2CC(CH3)3)4(tea)4(H2O)4]·4H2O (73, right)[61]


图 18 (左) [Zn2(L1)2DyCl3]·2H2O (76)、[Zn2(L1)2Dy(MeOH)Br3]·3H2O (77)、[Zn2(L1)2Dy(H2O)Br2][ZnBr4]0.5 (78)及[Zn2(L2)2DyCl3]·2H2O (79)的合成路线; (右)上左、上右、下左和下右分别代表76~79的磁滞回线[63]
Figure 18 (left) Synthesis procedure for [Zn2(L1)2DyCl3]·2H2O (76), [Zn2(L1)2Dy(MeOH)Br3]·3H2O (77), [Zn2(L1)2Dy(H2O)Br2][ZnBr4]0.5 (78) and [Zn2(L2)2DyCl3]·2H2O (79); (right) Magnetic hysteresis for 76 (top left), 77 (top right), 78 (bottom left) and 79 (bottom right)[63]


图 20 [ZnDy(LH4)2](NO3)3·6H2O (86)的晶体结构(左)[67]; 配合物[Zn4Dy2(L1)2(L2)2(N3)2]Cl2·2H2O(87)中L1、L2配体形成过程图(中)及其晶体结构图(右)[68]
Figure 20 Crystal structure (left) of [ZnDy(LH4)2](NO3)3·6H2O (86)[67]; Formation process (middle) of L1 and L2 in complex [Zn4Dy2(L1)2(L2)2(N3)2]Cl2·2H2O (87) and crystal structure of 87 (right)[68]

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:
 下载:
下载:
 下载:
下载: