College of Chemistry and Chemical Engineering, Hubei Key Laboratory of Pollutant Analysis & Reuse Technology, Hubei Normal University, Huangshi, Hubei 435002, China
2.
Institute for Advanced Materials of Hubei Normal University, Huangshi, Hubei 435002, China
Received Date:
25 March 2020 Revised Date:
02 June 2020 Available Online:
10 September 2020
Abstract:
The effect of ferroelectric polarization on the activity enhancement of Bi2MoO6 photocatalyst was explored by applying ferroelectric field polarization to organic-inorganic composite film materials constructed by ferroelectric photocatalyst Bi2MoO6 and polymethyl methacrylate (PMMA). The efficiency of unpolarized Bi2MoO6 degradation of rhodamine B (RhB) was 57.6% under 40 min light irradiation, and the degradation efficiency of bisphenol A (BPA) was 33.4% under 150 min light irradiation. The photocatalytic activity of Bi2MoO6 material polarized for 1.5 h at 15 V voltages was greatly enhanced, and the degradation efficiency of RhB and BPA under the same conditions reached 98.1% and 79.2%, respectively. The reason for the enhancement of photocatalytic activity is attributed to the enhancement of the internal electric field. The ferroelectric domains of the internal electric field of unpolarized Bi2MoO6 were disordered and unevenly distributed, and photogenerated carriers were very prone to recombination. When the applied electric field polarized Bi2MoO6, the ferroelectric domains of Bi2MoO6 tended to be ordered, and the polarization direction tended to be the same. Positive charges were generated on one side of the surface (C+ region) and negative charges were generated on the other side (C-region). The polarized electric field from C-region to C+ region drives the photogenerated electrons and holes to migrate to C+ and C-regions, respectively. This process promotes the rapid migration of photogenerated charge carriers from the interior to the surface, improves and prolongs the separation efficiency and lifetime of photogenerated carriers, leading to the enhancement of photocatalytic activity.
Figure 9.
UV-Vis absorption spectra of Bi2MoO6 (a) and BMO-2 (b) for degradation of BPA; Photocatalytic curves of BPA (c) and corresponding rate constant of the catalysts (d)
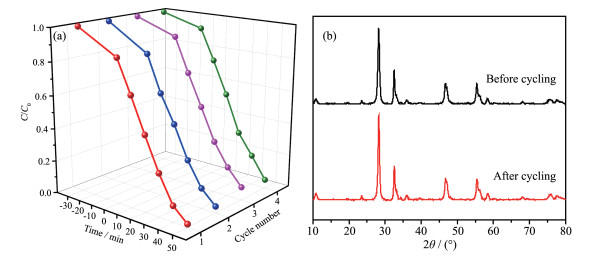
Figure 10.
Cycling performance for the photocatalytic degradation of RhB over Bi2MoO6 under visible light irradiation (a); XRD patterns before and after cycling experiment (b)
Figure 11.
UV-Vis DRS of Bi2MoO6, BMO-1, BMO-2 and BMO-3 (a); Plots of (αhν)2 versus hν for BMO and BMO-2 (b), and VB-XPS of Bi2MoO6 and BMO-2 (insert)
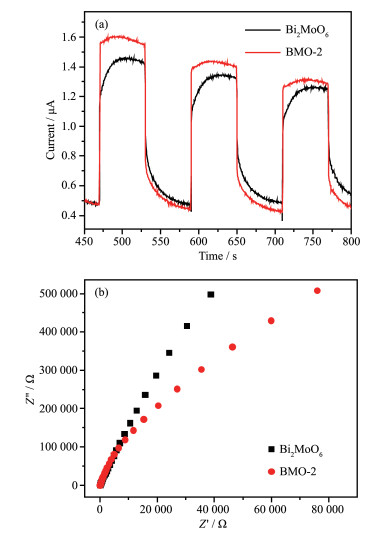
Figure 13.
Transient photocurrent responses for Bi2MoO6 and BMO-2 under visible light irradiation (λ>400 nm)
(a); Electrochemical impedance spectra of Bi2MoO6 and BMO-2 (b)
基于以上的表征分析,对极化后的Bi2MoO6提出了一种可能的光催化机理(图 15)。光催化活性提升的原因归因于内部电场的增强。未极化的Bi2MoO6的内电场的铁电畴是无序、分布不均匀的,光生载流子非常容易发生体内复合。当外加电场极化Bi2MoO6时,Bi2MoO6的铁电畴趋于有序,极化方向趋于同一方向,表面一侧(C+区)产生正电荷,在另一侧(C-区)产生负电荷,从C-区指向C+区的极化电场推动e-和h+分别迁移到C+和C-区域。这一过程促使光生电荷载流子快速从体内迁移至表面,提高和延长了光生载流子的分离效率和寿命,导致光催化活性的提升。Bi2MoO6的CB电位比O2/·O2-(0.28 eV vs NHE)的标准电位更负,这表明光生电子很容易将O2还原为·O2-[50]。然而,Bi2MoO6的VB电位下的空穴不能氧化水生成·OH,因为Bi2MoO6的标准电势相对H2O/·OH的标准电势(2.68 eV vs NHE)更负,所以·O2-进一步氧化H2O产生·OH。h+、·O2-和·OH氧化RhB或者BPA分子最终生成CO2和H2O,从而达到降解污染物的效果。
图 15
图 15.
BMO-2光催化剂降解污染物可能的机理
Figure 15.
Possible photocatalytic mechanism for photodegradation of pollutants over the BMO-2 photocatalyst
Figure 9
UV-Vis absorption spectra of Bi2MoO6 (a) and BMO-2 (b) for degradation of BPA; Photocatalytic curves of BPA (c) and corresponding rate constant of the catalysts (d)
Figure 10
Cycling performance for the photocatalytic degradation of RhB over Bi2MoO6 under visible light irradiation (a); XRD patterns before and after cycling experiment (b)
Figure 13
Transient photocurrent responses for Bi2MoO6 and BMO-2 under visible light irradiation (λ>400 nm)
(a); Electrochemical impedance spectra of Bi2MoO6 and BMO-2 (b)


 下载:
下载:













 下载:
下载:
















 下载:
下载:
