-
[1]
Dunn B, Kamath H, Tarascon J M. Science, 2011, 334(6058):928-935 doi: 10.1126/science.1212741
-
[2]
Ellis B L, Makahnouk W R M, Makimura Y, et al. Nat. Mater., 2007, 6(10):749-753 doi: 10.1038/nmat2007
-
[3]
Pan H L, Hu Y S, Chen L Q. Energy Environ. Sci., 2013, 6(8):2338-2360 doi: 10.1039/c3ee40847g
-
[4]
XIANG Xing-De(向兴德), LU Yan-Ying(卢艳莹), CHEN Jun (陈军). Acta Chim. Sinica(化学学报), 2017, 75: 154-162
-
[5]
Guo J Z, Yang Y, Wu X L, et al. Adv. Energy Mater., 2018, 8:1702504 doi: 10.1002/aenm.201702504
-
[6]
Kim H, Park I, Lee S, et al. Chem. Mater., 2013, 25(18):3614-3622 doi: 10.1021/cm4013816
-
[7]
Slater M D, Kim D, Lee E, et al. Adv. Funct. Mater., 2013, 23(8):947-958 doi: 10.1002/adfm.201200691
-
[8]
Vaalma C, Buchholz D, Weil M, et al. Nat. Rev. Mater., 2018, 3(4):18013 doi: 10.1038/natrevmats.2018.13
-
[9]
Yang X, Wang Y Y, Wu X L, et al. Acta Metall. Sin. (Eng. Lett.), 2020, DOI: 10.1007/s40195-020-01001-7
-
[10]
Kim S W, Seo D H, Ma X, et al. Adv. Energy Mater., 2012, 2(7):710-721 doi: 10.1002/aenm.201200026
-
[11]
Palomares V, Serras P, Villaluenga I, et al. Energy Environ. Sci., 2012, 5(3):5884-5901 doi: 10.1039/c2ee02781j
-
[12]
Gu Z Y, Guo J Z, Sun Z H, et al. Sci. Bull., 2020, 65(9):702-710 doi: 10.1016/j.scib.2020.01.018
-
[13]
Senthilkumar B, Murugesan C, Barpanda P, et al. Small Methods, 2019, 3(4):1800253 doi: 10.1002/smtd.201800253
-
[14]
Wang Q, Chu S Y, Guo S H. Chin. Chem. Lett., 2020, 10018417 https://doi.org/10.1016/j.cclet.2019.12.008.
-
[15]
Xu H, Guo S H, Zhou H S. J. Mater. Chem. A, 2019, 7:23662-23678 doi: 10.1039/C9TA06389G
-
[16]
Lu Y H, Wang L, Goodenough J B, et al. Chem. Commun., 2012, 48:6544-6546 doi: 10.1039/c2cc31777j
-
[17]
Hou B H, Wang Y Y, Wu X L, et al. ACS Appl. Mater. Interfaces, 2018, 10:3581-3589 doi: 10.1021/acsami.7b16580
-
[18]
Hou B H, Wang Y Y, Wu X L, et al. Adv. Funct. Mater., 2018, 28:1805444 doi: 10.1002/adfm.201805444
-
[19]
Liang H J, Hou B H, Wu X L, et al. Energy Environ. Sci., 2019, 12(12):3575-3584 doi: 10.1039/C9EE02759A
-
[20]
ZHANG Ning(张宁), LIU Yong-Chang(刘永畅), CHEN Jun (陈军). Chinese J. Inorg. Chem.(无机化学学报), 2015, 31(9): 1739-1750 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20150908&flag=1
-
[21]
Wang Y Y, Hou B H, Wu X L, et al. Adv. Energy Mater., 2018, 8:1703252 doi: 10.1002/aenm.201703252
-
[22]
Tripathi R, Ramesh T N, Ellis B L, et al. Angew. Chem. Int. Ed., 2010, 49:8738-8742 doi: 10.1002/anie.201003743
-
[23]
Lee K T, Ramesh T N, Nan F, et al. Chem. Mater., 2011, 23:3593-3600 doi: 10.1021/cm200450y
-
[24]
Kim J, Seo D H, Kim H, et al. Energy Environ. Sci., 2015, 8(2):540-545 doi: 10.1039/C4EE03215B
-
[25]
Wongittharom N, Wang C H, Chang J K, et al. ACS Appl. Mater. Interfaces, 2014, 6(20):17564-17570 doi: 10.1021/am5033605
-
[26]
Guo J Z, Wu X L, Wang R S, et al. Chem. Eur. J., 2015, 21:17371-17378 doi: 10.1002/chem.201502583
-
[27]
Ali G, Lee J H, Susanto D, et al. ACS Appl. Mater. Interfaces, 2016, 8(24):15422-15429 doi: 10.1021/acsami.6b04014
-
[28]
Liu Y C, Zhang N, Fan L Z, et al. Adv. Funct. Mater., 2018, 28(30):1801917 doi: 10.1002/adfm.201801917
-
[29]
Barpanda P, Liu G D, Ling C D, et al. Chem. Mater., 2013, 25(17):3480-3487 doi: 10.1021/cm401657c
-
[30]
Lin B, Zhang S, Deng C. J. Mater. Chem. A, 2016, 4(7):2550-2559 doi: 10.1039/C5TA09403H
-
[31]
Chen M Z, Chen L N, Dou S X, et al. Adv. Mater., 2017, 29(21):1605535 doi: 10.1002/adma.201605535
-
[32]
Singh P, Shiva K, Goodenough J B, et al. Energy Environ. Sci., 2015, 8(10):3000-3005 doi: 10.1039/C5EE02274F
-
[33]
Liu X H, Tang L B, Wang Y G, et al. Electrochim. Acta, 2019, 296:345-354 doi: 10.1016/j.electacta.2018.11.064
-
[34]
Chen M Z, Cortie D, Dou S X, et al. Adv. Energy Mater., 2018, 8(27):1800944 doi: 10.1002/aenm.201800944
-
[35]
Liu Y M, Rajagopalan R, Guo X D, et al. ACS Sustainable Chem. Eng., 2018, 6(12):16105-16112 doi: 10.1021/acssuschemeng.8b02679
-
[36]
Li S D, Guo J H, Yang Y, et al. ACS Appl. Mater. Interfaces, 2016, 8(27):17233-17238 doi: 10.1021/acsami.6b03969
-
[37]
Yao W J, Sougrati M T, Hoang K, et al. Chem. Mater., 2017, 29(21):9095-9101 doi: 10.1021/acs.chemmater.7b02764
-
[38]
Wu X H, Zhong G M, Yang Y. J. Power Sources, 2016, 327:666-674 doi: 10.1016/j.jpowsour.2016.07.061
-
[39]
Yuan T C, Wang Y X, Cao Y L, et al. Nano Energy, 2019, 56:160-168 doi: 10.1016/j.nanoen.2018.11.011
-
[40]
Pu X J, Wang H M, Cao Y L, et al. Energy Storage. Mater., 2019, 22:2405-8297
-
[41]
Chen M Z, Hua W B, Dou S X, et al. Nat. Commun., 2019, 10(1):1480 doi: 10.1038/s41467-019-09170-5
-
[42]
Brezesinski T, Wang J, Tolbert S H, et al. Nat. Mater., 2010, 9(2):146-151 doi: 10.1038/nmat2612
-
[43]
Baskar S, Angalakuthi R, Barpanda P, et al. ECS Trans., 2018, 85(13):227-234 doi: 10.1149/08513.0227ecst
-
[44]
Ma X D, Pan Z Y, Wu X H, et al. Chem. Eng. J., 2019, 365:132-141 doi: 10.1016/j.cej.2019.01.173
-
[45]
Rajagopalan R, Chen B, Dou S X, et al. Adv. Mater., 2017, 29(12):1605694 doi: 10.1002/adma.201605694
-
[46]
Shiva K, Singh P, Goodenough J B, et al. Energy Environ. Sci., 2016, 9(10):3103-3106 doi: 10.1039/C6EE01093H
-
[47]
Huang W F, Zhou J, Li B, et al. Sci. Rep., 2014, 4:4188
-
[48]
Deng X, Shi W X, Liu M L, et al. ACS. Appl. Mater. Interfaces, 2017, 9(19):16280-16287 doi: 10.1021/acsami.7b03933
-
[49]
Ling R, Cai S, Sun X H, et al. J. Alloys Compd., 2017, 704:631-640 doi: 10.1016/j.jallcom.2017.02.075
-
[50]
Li Q, Liu Z G, Yang Y, et al. Angew. Chem. Int. Ed., 2018, 57(37):11918-11923 doi: 10.1002/anie.201805555
-
[51]
Kundu D, Tripathi R, Popov G, et al. Chem. Mater., 2015, 27(3):885-891 doi: 10.1021/cm504058k
-
[52]
Recham N, Rousse G, Sougrati M T, et al. Chem. Mater., 2012, 24(22):4363-4370 doi: 10.1021/cm302428w
-
[53]
Liu D S, Liu D H, Wu X L, et al. Electrochim. Acta, 2018, 264:292-300 doi: 10.1016/j.electacta.2018.01.129
-
[54]
Liu Q N, Hu Z, Dou S X, et al. Small, 2019, 15(32):1805381 doi: 10.1002/smll.201805381
-
[55]
Wang P F, You Y, Guo Y G, et al. Adv. Energy Mater., 2018, 8(8):1701912 doi: 10.1002/aenm.201701912
-
[56]
Chen M Z, Liu Q N, Wang S W, et al. Adv. Energy Mater., 2019, 9(14):1803609 doi: 10.1002/aenm.201803609
-
[57]
Li X, Wang Y, Ceder G, et al. Chem. Mater., 2016, 28(18):6575-6583 doi: 10.1021/acs.chemmater.6b02440
-
[58]
Kikkawa S, Miyazaki S, Koizumi M. Mater. Res. Bull., 1985, 20(4):373-377 doi: 10.1016/0025-5408(85)90003-0
-
[59]
Zhao J, Zhao L, Okada S, et al. J. Electrochem. Soc., 2013, 160(5):A3077-A3081 doi: 10.1149/2.007305jes
-
[60]
Lee E, Brown D E, Alp E E, et al. Chem. Mater., 2015, 27(19):6755-6764 doi: 10.1021/acs.chemmater.5b02918
-
[61]
Xu J L, Han Z, Guo S H, et al. Small, 2020, 16:1904388 doi: 10.1002/smll.201904388
-
[62]
Li Y J, Gao Y R, Wang Z X, et al. Nano Energy, 2018, 47:519-526 doi: 10.1016/j.nanoen.2018.03.007
-
[63]
Gao Y R, Wang Z X, Lu G. J. Mater. Chem. A, 2019, 7(6):2619-2625 doi: 10.1039/C8TA10767J
-
[64]
Yabuuchi N, Kajiyama M, Komaba S, et al. Nat. Mater., 2012, 11(6):512-517 doi: 10.1038/nmat3309
-
[65]
Mortemard de Boisse B, Carlier D, Guignard M, et al. J. Electrochem. Soc., 2013, 160(4):A569-A574 doi: 10.1149/2.032304jes
-
[66]
Li W H, Ning Q L, Wu X L, et al. Adv. Mater., 2019, 31:1804766 doi: 10.1002/adma.201804766
-
[67]
Zhou S, Wang D W. ACS Nano, 2010, 4(11):7014-7020 doi: 10.1021/nn102194w
-
[68]
Ge J P, Zhang Q, Yin Y D, et al. Angew. Chem. Int. Ed., 2008, 47(46):8924-8928 doi: 10.1002/anie.200803968
-
[69]
Kalluri S, Seng K H, Dou S X, et al. RSC Adv., 2013, 3(48):25576-25601 doi: 10.1039/c3ra45414b
-
[70]
WANG Ling(王玲), YANG Guo-Rui(杨国锐), YAN Wei(延卫), et al. Acta Chim. Sinica(化学学报), 2018, 76: 259-277
-
[71]
Kalluri S, Seng K H, Dou S X, et al. ACS Appl. Mater. Interfaces, 2014, 6(12):8953-8958 doi: 10.1021/am502343s
-
[72]
Zhao W W, Tsuchiya Y, Yabuuchi N. Small Methods, 2018, 3(4):1800032
-
[73]
Yoshida H, Yabuuchi N, Komaba S. Electrochem. Commun., 2013, 34:60-63 doi: 10.1016/j.elecom.2013.05.012
-
[74]
Kubota K, Asari T, Komaba S, et al. Adv. Funct. Mater., 2016, 26(33):6047-6059 doi: 10.1002/adfm.201601292
-
[75]
Nanba Y, Iwao T, Yamada A, et al. Chem. Mater., 2016, 28(4):1058-1065 doi: 10.1021/acs.chemmater.5b04289
-
[76]
Hasa I, Buchholz D, Passerini S, et al. Adv. Energy Mater., 2014, 4(15):1400083 doi: 10.1002/aenm.201400083
-
[77]
Lu Z, Dahn J R. J. Electrochem. Soc., 2002, 149(7):A815-A822 doi: 10.1149/1.1480014
-
[78]
Yuan D D, Hu X H, Cao Y L, et al. Electrochim. Acta, 2014, 116:300-305 doi: 10.1016/j.electacta.2013.10.211
-
[79]
Buchholz D, Chagas L G, Vaalma C, et al. J. Mater. Chem. A, 2014, 2(33):13415-13421 doi: 10.1039/C4TA02627F
-
[80]
Duffort V, Talaie E, Black R, et al. Chem. Mater., 2015, 27(7):2515-2524 doi: 10.1021/acs.chemmater.5b00097
-
[81]
Mu L, Xu S, Hu Y S, et al. Adv. Mater., 2015, 27(43):69286933
-
[82]
Li Y M, Yang Z Z, Hu Y S, et al. Adv. Sci., 2015, 2(6):1500031 doi: 10.1002/advs.201500031
-
[83]
Vassilaras P, Toumar A J, Ceder G. Electrochem. Commun., 2014, 38:79-81 doi: 10.1016/j.elecom.2013.11.015
-
[84]
Li X, Wu D, Ceder G, et al. Electrochem. Commun., 2014, 49:51-54 doi: 10.1016/j.elecom.2014.10.003
-
[85]
Hwang J Y, Myung S T, Sun Y K. J. Phys. Chem. C, 2018, 122(25):13500-13507 doi: 10.1021/acs.jpcc.7b12140
-
[86]
Keller M, Buchholz D, Passerini S. Adv. Energy Mater., 2016, 6(3):1501555 doi: 10.1002/aenm.201501555
-
[87]
Jiang X L, Hu F D, Zhang J S. RSC Adv., 2016, 6(105):103238-103241 doi: 10.1039/C6RA22818F
-
[88]
Liu Y, Qiao Y, Zhang W X, et al. Nano Energy, 2015, 12:386-393 doi: 10.1016/j.nanoen.2015.01.012
-
[89]
Wang L, Lu Y, Goodenough J B, et al. Angew. Chem. Int. Ed., 2013, 52(7):1964-1967 doi: 10.1002/anie.201206854
-
[90]
Wang L, Song J, Qiao R M, et al. J. Am. Chem. Soc., 2015, 137(7):2548-2554 doi: 10.1021/ja510347s
-
[91]
Jiang Y Z, Yu S L, Dou S X, et al. Adv. Funct. Mater., 2016, 26(29):5315-5321 doi: 10.1002/adfm.201600747
-
[92]
Tan S Y, Wang L, Bian L, et al. J. Power Sources, 2015, 277:139-146 doi: 10.1016/j.jpowsour.2014.11.149
-
[93]
You Y, Yao H R, Guo Y G, et al. Adv. Mater., 2016, 28(33):7243-7248 doi: 10.1002/adma.201600846
-
[94]
Shi N K, Gao Q L, Chen J, et al. Dalton Trans., 2019, 48(11):3658-3663 doi: 10.1039/C8DT05111A
-
[95]
Zhang L J, Ji S M, Liu J, et al. RSC. Adv., 2017, 7(39):2400424010
-
[96]
Zhao S, Li Y, Yang Z H, et al. ACS Appl. Mater. Interfaces, 2019, 11(19):17425-17434 doi: 10.1021/acsami.9b03077
-
[97]
CAO Dun-Ping(曹敦平), YIN Cong-Ling(尹从岭), LI ChiLin(李驰麟), et al. Chin. Sci. Bull.(科学通报), 2017, 62(9): 897-907
-
[98]
Liu D H, Li W H, Wu X L, et al. Adv. Mater., 2018, 30:1706317 doi: 10.1002/adma.201706317
-
[99]
Wang Y Y, Fan H, Wu X L, et al. J. Mater. Chem. A, 2018, 6:22966-22975 doi: 10.1039/C8TA09264H

 下载:
下载:
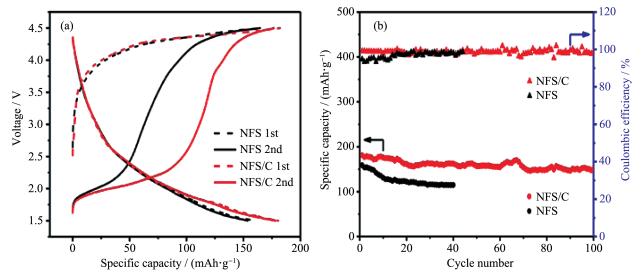
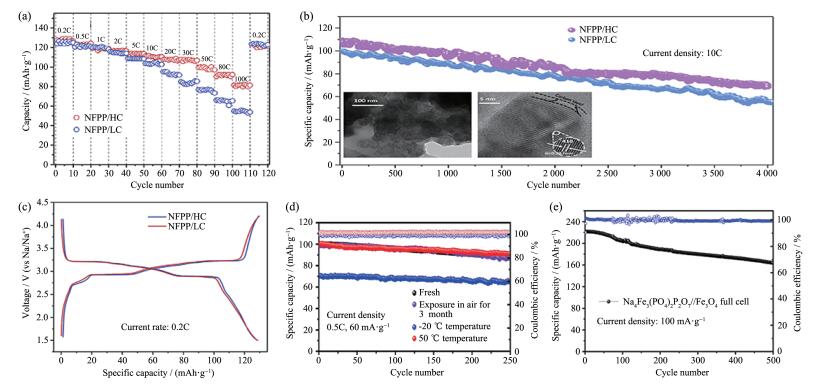
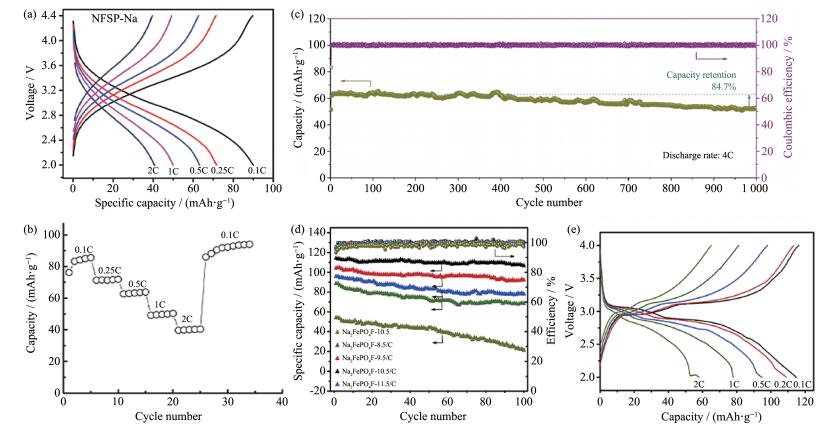
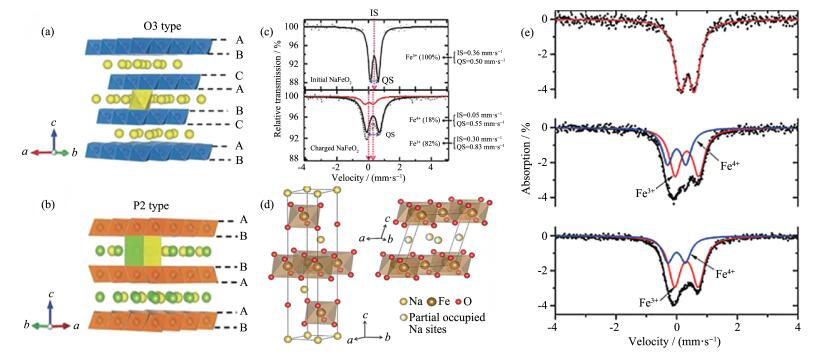



 下载:
下载:














 下载:
下载:
