Received Date:
16 December 2019 Revised Date:
28 February 2020 Available Online:
10 May 2020
Abstract:
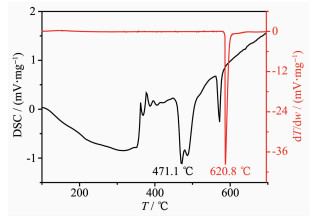
In this paper, an ordered mesoporous Si/C composite structure was prepared successfully for the anode materials of Li-ion batteries through the synthesis of SiO2-ordered mesoporous precursor (SiO2-OMPs) by a P123-copolymer templet route and then a magnesiothermic reduction reaction (MRR) for the SiO2-OMPs precursor and ensuing a phenolic-resin based carbon-capsuling treatment. From the observations of SEM/TEM, it is found that the morphology of SiO2-OMPs can be got well-controlled by HCl concentration and obtained a short rod with densified geometrical stacking under high acid concentration, and the ordered mesoporous symmetry kept unchanged perfectly through later MRR and C-capsuling routes. In addition, the detailed and reasonable analyses based on XRD data reveal that MRR process consists of two-step serial reactions:the Mg2Si intermediate formation from Mg and SiO2 reactants and its transition to final Si products by re-reducing SiO2. The slow feature of the solid/solid diffusive reaction in the second step restrains the completion of whole reduction process, resulting in a low yield of silicon and the existence of some impurity phases. Electrochemical measurements show that the ordered mesoporous Si/C composite exhibited excellent cyclic stability and rate performance, which can be attributed to the robust architect and unblocked mesopore system within such composite structure.
Figure 1.
Mesostructure and mesopore analyses for as-synthesized Si02-0MPs samples SI and S2; (a) SAX patterns and(b) HRTEM image for sample SI; (c) N2 adsorption-desorption isotherms and (d) pore-size distribution diagrams for samples SI and S2
Figure 2.
FESEM images of as-synthesized (a) S1 and (b, c) S2 samples and (d) variations of length and aspect ratio of as-synthesized SiO2-OMPs with concentration of HCl
Figure 4.
XRD patterns of products obtained by MRR: (a) Reduction for 5 h at 400, 560, 600, 620 and 660 C; (b) Reduction for 5, 10, 15 and 20h at 560 C
Figure 7.
Electrochemical measurements for as-prepared Si-OMPs and Si/C-OMPs samples: V-Q curves for the first 10 cycles; Cycled curves at a current rate of0.1 A.g-1; (c) Varied-curent rate cycling curves
Figure 1
Mesostructure and mesopore analyses for as-synthesized Si02-0MPs samples SI and S2; (a) SAX patterns and(b) HRTEM image for sample SI; (c) N2 adsorption-desorption isotherms and (d) pore-size distribution diagrams for samples SI and S2
Figure 2
FESEM images of as-synthesized (a) S1 and (b, c) S2 samples and (d) variations of length and aspect ratio of as-synthesized SiO2-OMPs with concentration of HCl
Figure 7
Electrochemical measurements for as-prepared Si-OMPs and Si/C-OMPs samples: V-Q curves for the first 10 cycles; Cycled curves at a current rate of0.1 A.g-1; (c) Varied-curent rate cycling curves


 下载:
下载:






 下载:
下载:








 下载:
下载:
