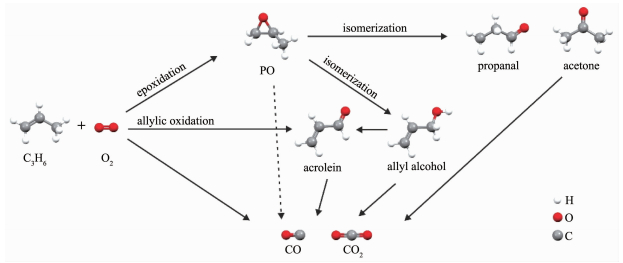
图 1.
丙烯氧化的反应网络
Figure 1.
Reaction network for the oxidation of C3H6 by molecular O2

环氧丙烷(propylene oxide,PO)是一种非常重要的化工原料,主要用于生产聚醚多元醇、丙二醇、表面活性剂等化学品。2013年全世界PO的年生成量是800万吨,并以每年4%~5%的速度增长[1]。目前工业上生产PO的方法主要有氯醇法、共氧化法(halcon)和以H2O2为氧化剂的部分氧化法(hydrogen peroxide-propylene oxide,HPPO),它们分别占PO总产能的43%、48%、5%,其余方法约占4%[2]。然而,这些方法面临着成本高、副产物多以及污染严重等问题,比如氯醇法使用Cl2作为原料会产生大量高pH值的含Cl废水和废渣等,而HPPO法因为使用昂贵的H2O2作为原料导致生产成本过高[2-3]。使用氧气和丙烯进行反应制备PO的直接氧化法(direct epoxidation of propylene by molecular O2,DEP),是理论上最为理想的PO生产方式(C3H6+1/2O2→C3H6O),具有原子经济、环保、低能耗等优点。但是,丙烯分子中活泼的α-H在反应过程中很容易发生脱除生成丙烯醛,成为环氧化最主要的竞争反应。另外,产物PO分子具有很高的反应活性[4],很容易发生异构化生成丙烯醇、丙醛、丙酮等,并在O2的作用下最终生成CO和CO2,如图 1所示。因为DEP反应过程中存在这些特点,也被誉为最具挑战的催化反应之一[5]。
早期研究者主要集中于Au和Ag催化剂催化丙烯环氧化反应的研究[6-7],然而对于只有丙烯和O2参与的DEP反应,Au和Ag的催化性能并不好。21世纪初期,Lambert等[8-14]发现Cu比Ag具有更优异的烯烃环氧化反应性能,并引发了国际上研究Cu基催化剂的热潮。近十几年来,已有一些关于丙烯环氧化反应的综述[1-2, 15-17],但主要针对Au、Ag等催化剂体系,而对Cu基催化剂的进展缺乏系统的评述。本综述归纳了近十几年来使用Cu基催化剂进行DEP反应的研究结果,集中于讨论Cu基催化剂提高DEP催化性能的改性策略,并对其仍然面临的问题和挑战进行总结。其中,Cu基催化剂的改性方法主要从晶面效应、双金属改性、载体效应、金属氧化物以及助剂修饰等方面进行论述。本综述充分展示Cu基催化剂的发展现状,为深层次揭示反应机理和高效DEP催化剂的开发提供参考。
目前为止,最受关注的DEP反应催化剂主要是集中在Au、Ag、Cu三类币族金属催化剂。Haruta课题组[18-20]开发了在H2辅助下使用Au催化剂进行丙烯环氧化反应的方法,并表现出较好的催化性能,但是在没有H2和水汽时,Au对PO没有任何选择性[16, 21]。Ag基催化剂已应用于工业乙烯环氧化反应,故倍受研究者的关注。但是对于DEP反应,Ag基催化剂表面深度氧化产物的速率比环氧化产物的生成速率高了约2个数量级[6],导致了Ag催化剂在DEP反应中表现不佳。
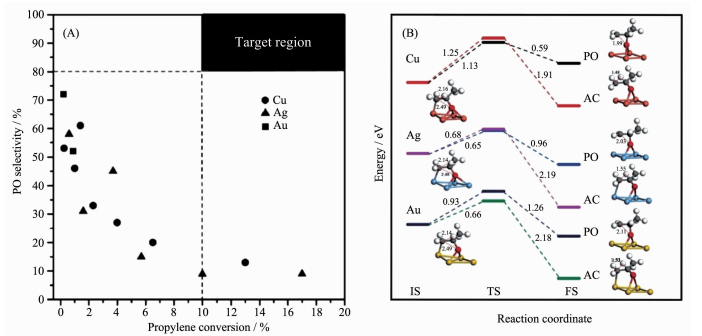
在高真空表面科学研究的过程中,Lambert等[13]发现Cu在烯烃环氧化过程中具有较好的催化选择性,并在2005年报道了将1% Cu负载在SiO2上进行常压DEP反应,在225 ℃时可以得到高达53%的PO选择性和0.25%的丙烯转化率。理论计算表明在Cu(111)上吸附的O原子碱性比Ag(111)上的更低,这使得丙烯环氧化过程中丙烯分子更容易在Cu表面形成丙烯金属氧环中间体(propylene oxametalla-cycle,OMMP)并最终转化为PO,而Ag(111)更容易导致α-H脱除产生丙烯醛和深度氧化产物[14]。2012年,Haruta等[16]对比了DEP反应中Au、Ag和Cu的催化性能,发现整体上Cu基催化剂相对于其他两者具有较好的表现,如图 2A所示。Dai等[22]的理论计算也支持了这个观点,发现在Cu(111)表面金属氧环中间体异构化为PO的能垒比异构化为丙酮更有利,而对于Ag(111)和Au(111)表面则不可避免地形成丙酮(图 2B)。
此外,Cu比Au和Ag的价格要低很多,这使得Cu催化剂在工业成本上有很大的优势。但是,未经改性的Cu催化剂仍远未达到工业化的目标,而且在催化反应过程中很容易发生团聚、氧化而导致失活。因此,需要通过调变载体、助剂、多组分组合等方式对Cu基催化剂进行改性以提高其催化性能和稳定性。
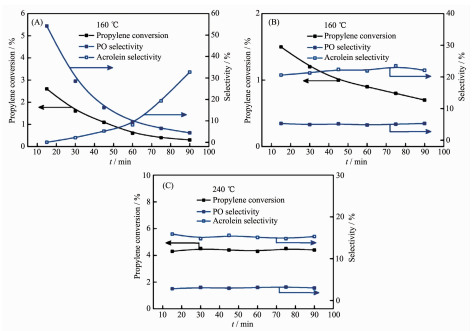
在DEP反应中,Cu基催化剂的价态和形貌结构受原料气中丙烯和氧的比例、水蒸气的压力、反应温度等多种因素影响。Cu基催化剂的不稳定性导致其在DEP反应过程中活性相的鉴定成了一个难点[23]。目前为止,Cu0、Cu+和Cu2+均有被认为是DEP活性相的报道,并给出了相对应的表征和分析。Vaughan等[13]通过检测Cu的俄歇谱等认为在SiO2载体上高分散的Cu0是丙烯环氧化的活性相,而Cu2O相则催化丙烯生成丙烯醛,CuO相会导致丙烯深度氧化。Marimuthu等[24]也认为Cu0比Cu2O具有更高的PO选择性。他们发现当有光照时,Cu@Cu2O上PO的选择性会从20%左右迅速升高到接近50%,其原因可能是光照引起的局域表面等离子共振(LSPR)作用使得表层的Cu2O被还原成Cu。Zheng等[25]对比了不同价态的Cu催化剂的DEP反应,发现对于PO的选择性Cu基催化剂按Cu > Cu2O > CuO顺序递减。同时,他们也发现Cu0在反应条件中很容易被氧化变成Cu+,故在反应刚开始时Cu0是活性相,反应稳定后Cu+是活性相,而CuO需要在更高的温度下(240 ℃)才有反应活性,如图 3所示。Su等[26]在碱金属修饰的Cu/SiO2体系研究中,通过红外表征认为Cu0和Cu+是活性相,而Cu2+容易产生丙烯醛和COx。
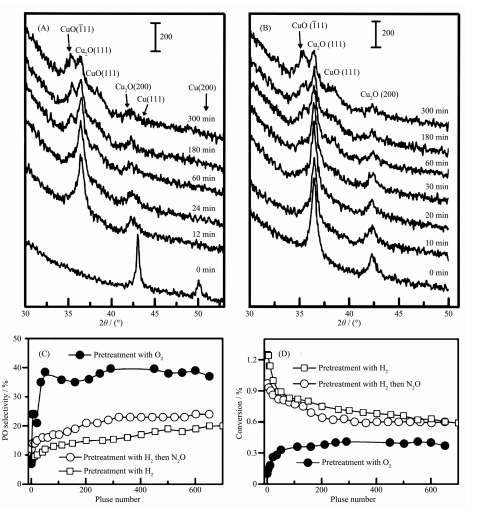
也有一些研究者认为反应条件下Cu+才是真正的活性相。比如,Zhu等[27]对K+修饰的CuOx-SiO2催化剂进行H2预处理,通过CO吸附红外和XRD研究发现预处理后的Cu0在丙烯环氧化反应气氛中会被氧化成Cu+,此时PO的生成速率极大地提高。He等[4]在5% CuOx/SiO2和Cs+-5% CuOx/SiO2体系中,通过原位XRD和FT-IR发现O2预处理得到的Cu2+作为初始材料在反应气氛中会出现Cu+,通过H2预处理得到的Cu0作为初始材料在反应气氛中会形成Cu+和Cu2+共存的混合物,而通过N2O预处理得到的Cu+作为初始材料在反应气氛中会出现Cu2+,如图 4A和4B所示。如果用脉冲法向3种不同方式预处理的催化剂中引入丙烯和O2,可以观察到随着脉冲数量增加催化剂将主要以Cu+的形式存在。图 4C和4D显示了脉冲过程中催化剂的性能,充分说明了Cu+对PO生成的重要作用。为了验证Cu+和Cu2+的丙烯环氧化催化活性,Düzenli等[28]运用密度泛函理论(density functional theory,DFT)对CuO(001)和Cu2O(001)的DEP反应过程进行了细致计算,发现相比于CuO(001),Cu2O(001)表面的吸附丙烯发生α-H脱除后的中间态形成丙烯醛过渡态需要克服0.87 eV的能垒,而Cu2O(001)比CuO(001)形成PO所需要克服的能垒低很多,故Cu2O(001)面上可选择性形成PO。但是,Seubsai等[29]认为在RuO2-CuO-NaCl/SiO2反应体系中,CuO是形成环氧丙烷的关键活性位点,它会与RuO2协同完成丙烯到PO的转化。
综上所述,大部分研究者认为Cu基催化剂中的Cu0和Cu+是DEP的活性相,而Cu2+不利于环氧化形成PO。由于未修饰的Cu基催化剂对DEP反应的活性和选择性都不高,因此在研究具体活性位时如果只看Cu的价态而不综合考虑Cu的化学环境是不合理的。
近年来使用表面结构明确的模型催化剂进行催化机理研究成了一个热门方向,它使得研究工业催化条件下的催化剂构效关系变成可能,并且在理论计算的帮助下可以从分子、原子层面认识和理解催化反应机理[30]。本课题组通过热力学和动力学参数调控制备了一系列特定表面结构的纳米晶[31],在催化反应过程中我们发现以高能晶面裸露的纳米晶表现出更加优异的催化性能[32-34]。比如,以高能晶面(332)裸露的Cu2O纳米晶相对于以(111)和(100)晶面裸露的Cu2O纳米晶表现更高的CO催化活性[34]。然而,用特定晶面的Cu基催化剂进行DEP反应机理研究的报道现在还比较少。
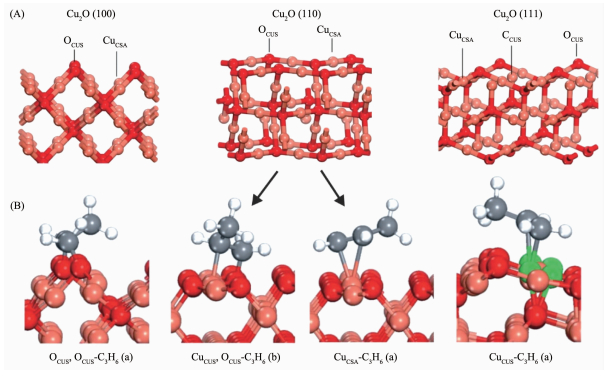
Hua等[35]用湿化学法制备了不同晶面裸露的Cu2O微纳米颗粒,并直接作为催化剂进行DEP反应,不同晶面对应的催化性能表现出较大的差距。如图 5所示,以(111)晶面裸露的八面体催化活性最高,同时对丙烯醛的选择性最高为70%左右;以(100)晶面裸露的立方体对CO2的选择性最高为80%左右;在3个样品中,以(110)晶面裸露的菱形十二面体对PO的选择性最高为20%左右。通过漫反射傅立叶变换红外光谱测试和DFT计算发现Cu2O(111)表面一配位的CuCUS、Cu2O(110)表面三配位的OCUS以及Cu2O(100)表面两配位的OCUS分别是丙烯醛、PO和CO2的活性位点,如图 6所示。丙烯吸附到菱形十二面体Cu2O(110)表面存在两种情况,即丙烯分子中的C=C双键同时吸附到单个Cu上以及分别吸附到Cu和周围不饱和配位的O上,后者在能量上更有优势,同时后者形成C3H6O(a)所需要的能量比C=C双键断裂低0.80 eV,解释了为什么Cu2O(110)晶面具有更高的PO选择性。Düzenli等[28]发现在Cu2O(001)上吸附的丙烯异构化为OMMP放热0.69 eV,OMMP转化成PO过渡态需要克服0.74 eV的能垒,PO脱附需要克服0.78 eV的能垒。不过,吸附的丙烯发生α-H脱除过程放热2.25 eV,形成丙烯醛过渡态要克服0.87 eV的能量,丙烯醛脱附需克服0.74 eV的能量。总体上Cu2O(001)更倾向于形成丙烯醛,这个结果与图 5A的性能表征结果相符,丙烯醛的选择性高于PO。

需要指出的是,Hua等的理论计算仅停留在形成OMMP中间态的动力学能垒这一步,并且没有考虑O2活化及其影响。而在之前的工作中,Lambert等[14]认为吸附态的原子氧Oa是在Cu(111)上形成PO的关键。Song等[36]计算了在有氧气参与的情况下Cu2O(111)进行DEP反应的动力学过程,研究发现吸附分子O2-具有相对低的碱性表现出最高的环氧化选择性,而吸附原子Oa具有最高的碱性主要生成丙烯醛,晶格氧O2-则反应活性很低。除此之外,中空结构催化剂能够提高材料利用率、增加比表面积进一步提高催化性能,比如我们在无表面活性剂条件下合成了中空的Cu2O纳米立方体[37],在DEP反应中其表现出比实心立方体更优异催化性能[38]。因此,对于深入理解Cu基催化剂的构效关系还需更加系统和细致的研究。
对于金属催化剂,引入异种金属是一种常见的调变策略。双金属可形成异质结构、核壳结构、固溶体、或金属间化合物等,均可有效地调节催化剂整体的形貌、尺寸和结构[39-40]。例如,不同金属间电负性差异会导致电子转移,不同金属的晶格失配导致晶格应力等,这些效应对催化性能可能产生很大的影响[41-42]。Zheng等[43]发现Ag95-Cu5/BaCO3体系表现出较好的催化性能,在丙烯转化率为3.6%时可以得到55.1%的PO选择性。这是因为Cu电负性(1.90)比Ag(1.93)低,在形成双金属催化剂时将略带正电,有利于稳定亲电的氧物种。另一个有趣的发现是,形成的Ag-Cu颗粒结构较为稳定,不易发生团聚,从而可以提高催化的稳定性[25]。
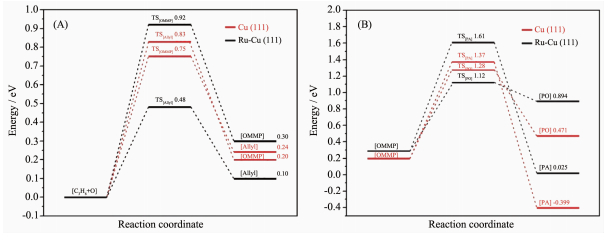
此外,Chimentao等[44]通过调节Au-Cu合金的比例发现提高Cu含量会导致颗粒尺寸变小,并且会影响催化剂在TiO2载体上的分散度。在XPS研究发现Cu会使Au的4f谱峰朝低结合能端位移,说明部分电子从Cu转移到了Au上,在丙烯环氧化反应中表现出40.5%的PO选择性。Kizilkaya等[45]通过理论计算发现如果在Ru的上面放置一层Cu(111)形成Ru-Cu(111)界面,Ru的部分电子会转移到Cu上导致吸附在Cu上的O更为亲核,从而使得α-H更加容易脱除导致PO选择性下降。图 7对比了Cu(111)和Ru-Cu(111)丙烯环氧化的反应能垒,可以看出Cu(111)对生成PO更有优势。此外,在催化剂制备过程中需要特别注意反应气氛和温度引起的合金化以及去合金化问题,比如在H2气氛中处理时Cu和Au会发生合金化现象形成合金,而在高温煅烧时则会发生去合金化现象[46]。
大部分的工业催化剂需要分散到载体上进行结构支撑,载体的锚定作用还可以抑制反应过程中的团聚现象,提高催化剂的分散度和稳定性。同时,一些载体还可以与催化剂发生电子相互作用,改善催化剂的电子结构以提高催化性能。在DEP反应中,Cu基催化剂最常使用的载体是SiO2、α-Al2O3、分子筛等。Duzenli等[47]将Cu负载到两种不同比表面积的无定形SiO2载体上,研究发现大比表面积的SiO2上具有较高的PO生成速率。Wang等[27]采用具有大比表面的SBA-15分子筛作为载体制备了1% CuOx/SBA-15催化剂,其PO的生成速率为2.1 mmol·g-1·h-1,反应的TOF是17.5 h-1。上述工作表明,大比表面积的载体更有利于Cu基催化剂的分散和稳定,故可提高催化性能。
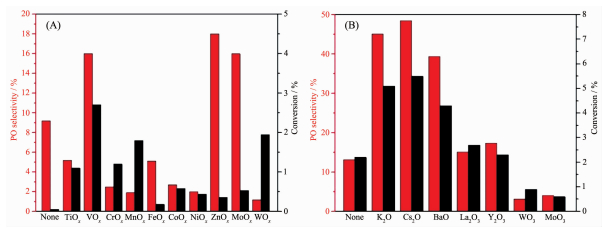
另一方面,金属氧化物修饰同样可以起到稳定Cu基催化剂和降低O2的吸附活化能的作用,使其在DEP反应中表现出更优异的性能。比如,Yang等[48]用TiOx、VOx、MnOx、FeOx、ZnOx、MoOx和WOx等过渡金属氧化物对Cu进行修饰,结果显示VOx、ZnOx和MoOx均有较好的DEP催化增强作用,如图 8A所示。其中,对于VOx-Cu催化剂,V与Cu的比例为0.11时表现出最好的DEP催化性能,丙烯转化率为2.7%和PO选择性为16%。通过实验表征和分析发现VOx有利于Cu的分散,并且能够促进Cu0转变成Cu2O进而提高催化性能,但是过量的VOx会产生强的酸位点而引起PO异构化导致副产物的生成。Zheng等[25]在Ag8Cu1/α-Al2O3体系的基础上引入金属氧化物形成Ag8Cu1/0.1% MOx/α-Al2O3多组分结构,研究发现DEP反应性能能够进一步得到优化,如图 8B所示。Seubsai等[49-53]在CuO-NaCl/SiO2体系中加入金属氧化物构建了RuO2-CuO-NaCl/SiO2、SnO2-CuO-NaCl/SiO2、Sb2O3-CuO-NaCl/SiO2和RuO2-CuO-NaCl-TeO2-MnOx/SiO2等体系,使得PO的生成速率得到了很大的提升。
添加助剂是工业催化剂中常用的改性策略。一般认为助剂可提高催化剂的活性、选择性和稳定性。例如,在工业化的乙烯环氧化反应中Cs和Cl的加入可以提高对目标产物的选择性[54]。对于烯烃环氧化反应来说,最常用的助剂是碱金属、碱土金属和卤素等三大类助剂。碱金属和碱土金属可能起到以下几个方面的作用:降低CuOx的尺寸,提高其分散性;稳定CuOx催化剂结构,提高催化稳定性;降低CuOx的路易斯酸性,抑制PO异构化形成丙烯醇进而氧化成丙烯醛;降低晶格氧的反应活性,抑制丙烯醛的产生[4, 26]。不过,针对不同的体系其具体作用会有所差异。
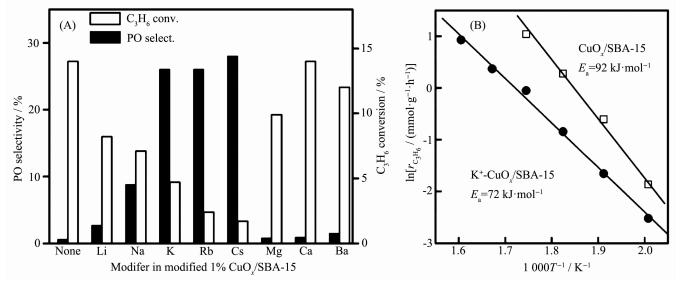
碱金属和碱土金属是DEP反应最常用的助剂。Wang等[27]使用Na、K、Cs、Mg、Ca等修饰CuOx/SBA-15催化剂,发现K、Rb和Cs表现出较好的改性效果,能将PO的选择性从接近0提升到25%以上,如图 9所示。其中,K+修饰的样品的PO选择性最高可超过50%,生成速率最高可达到2.2 mmol·g-1·h-1。动力学计算发现K+加入后DEP反应的活化能从92 kJ·mol-1降到72 kJ·mol-1(图 9B)。Su等[26]也发现K+能够显著提高Cu/SiO2对PO的选择性,其中KAc比KCl具有更高的PO选择性,能达到60%左右,很低转化率时甚至可以接近100%。同样地,碱金属特别是Cs+修饰的5% CuOx/SiO2能够极大地提高PO选择性,从1.8%提高到34%,PO产率提高了将近10倍,从0.27%提升到2.6%[4]。Teran等[55]发现碱金属K+、Na+可以减少CuOx中O的亲核性质而降低α-H脱除的倾向,提高PO选择性,而碱土金属Ca2+会吸附-OH而导致CuOx发生电荷补偿,因此只能提高催化活性和稳定性,对催化选择性没有贡献。通过以上研究可以发现碱金属和碱土金属会影响Cu基催化剂吸附O的电子性质,可以在一定程度上提高Cu基催化剂的DEP反应性能,不过过量的修饰会导致催化活性位点被覆盖而发生催化剂毒化现象。
卤素(Cl或Br)也是烯烃环氧化催化剂中常用的助剂,一般认为其可以改善表面吸附O的亲电性。Seubsai等[56]考察了RuO2-CuOx-NaCl/SiO2催化剂的DEP反应性能,发现引入微量的含氯碳氢化合物(比如二氯乙烷)可以补充催化剂表面Cl的流失从而稳定对环氧丙烷的选择性。同时,Cl或者含Cl化合物会毒化催化剂表面的活性位点使有效的O2吸附位点减少,导致丙烯的转化率下降。Zhang等[57]改变Ag-Cu-Cl/BaCO3催化剂中Cl的修饰量发现适量的Cl负载有助于提高PO选择性,能够降低吸附O2分解为O原子基团而导致丙烯完全燃烧的倾向,在200 ℃时PO的选择性能够达到83.7%。
也有一小部分研究者认为Cl对烯烃环氧化反应的贡献其实很小。Vaughan等[13]在Cu/SiO2催化剂上负载NaCl和NH4Cl发现它们都不利于PO的生成。在大多数研究体系中,Cl被认为能有效地降低Cu基催化剂表面吸附O的碱性而提高对环氧产物的选择性,但是需要对Cl的修饰量进行控制,过量的Cl会导致催化剂中毒使得催化性能恶化。不过,目前大部分体系过于复杂,在众多的影响因素中对Cl的机理进行解释显得不够令人信服,因此还需要在更加明确的体系中对其机理进行深入研究。
通过传统外负载方式制备的催化剂很容易在催化过程中发生Cl的流失,导致选择性下降。Seubsai等[58]在RuO2-CuO/SiO2负载NaCl发现NaCl能够显著提高PO的生成速率,但是其稳定性很差,从开始的生成速率1 336 gPO·h-1·kgcat-1快速下降,4 h后催化剂完全失活。同时,他们将Ru-Cu-Na/SiO2催化剂事先通含有水汽的N2进行处理,然后与未处理的催化剂进行催化稳定性测试,发现事先预处理过的样品比未处理的性能差很多。通过对XPS、EDS和ICP-OES的综合分析发现在催化过程中Cl发生了流失,NaCl转化为没有活性的Na2O。在环氧乙烷的工业化生产中,需要持续通入含Cl添加剂以补充反应过程中Cl的流失,以维持反应的稳定性。如果催化剂中Cl结构能够稳定存在就可以得到高的稳定性和催化性能,提高工业效率,并且能减少工业成本。然而,目前对于Cu基催化剂还没有能有效稳定Cl的策略。
综上所述,相对于目前工业化的PO生产方法,DEP反应拥有工艺简单、环境友好和原子经济等优点,是最为理想的生产方式。但是,丙烯分子中活泼的α-H和PO的高活性使得PO的选择性比较低,Cu基催化剂表面吸附O的碱性比Au和Ag基催化剂的更低这使得它对提高PO的选择性更有优势而受到了广泛关注。近十几年来,研究者通过载体、助剂、晶面控制和多组分组合的方式对Cu基催化剂进行改性,取得了许多进展。表 1总结了近年来具有代表性的Cu基催化材料的反应条件和催化性能。
 下载:
导出CSV
下载:
导出CSV
| nC3H6/nO2 | Catalyst | Conv./% | Se1ect./% | PO formation rate / (μmo1·g-1·h-1) | T0F/ h-1 | Temp./℃ | Space ve1ocity / h-1 | Pressure/MPa | Ref. |
| 10/1 | VCe0.2Cu0.8-NaC1 | 0.19 | 43.4 | 162.5 | — | 250 | — | 0.1 | [59] |
| 1/2 | Ag-CuC1 | 2.33 | 33.6 | — | — | 350 | 18 000 | 0.1 | [60] |
| 1/1 | 1%Cu/SiO2 | 0.25 | 53 | — | 0.09 | 225 | 30 000 | 0.1 | [13] |
| 1/1 | K+-1% CuOx/SBA-15 | 0.7 | 41 | 2 100 | 17.5 | 350 | 18 000 | 0.1 | [5] |
| 6/5 | K+-1% CuOx/SBA-15 | 2.2 | 24 | 2 200 | 18 | 350 | 18 000 | 0.1 | [27] |
| 1/1 | K+-5% CuOx-SiO2 | 2.0 | 25 | 2 000 | — | 275 | 18 000 | 0.1 | [27] |
| 1/1 | KAc-Cu/SiO2 | 1.3 | 61 | — | — | 275 | 30 000 | 0.1 | [26] |
| 2/1 | VOx-Cu | 2.7 | 16 | 230 | — | 230 | 15 000 | 0.1 | [48] |
| 22/9 | CuAuDSiO2 | 0.13 | 70 | — | — | 300 | 22 500 | 0.1 | [46] |
| 4/1 | RuO2-Cu0-NaC1/Si02 | 10 | 50 | — | — | 250 | 20 000 | 0.1 | [49] |
| 1/2 | 3%Cu-2.25%K/m-SiO2 | 2.90 | 20.49 | 1 549 | — | 350 | 20 000 | 0.1 | [47] |
| 1/39.5 | RuOx-CuOx/SiO2 | 13 | 26 | — | — | 225 | 18 000 | 0.1 | [61] |
| 2/1 | Ag-CuDBaCO3 | 3.6 | 55.1 | — | — | 200 | 2 000 | 0.1 | [43] |
| 1/39.5 | Cs+-5%CuOx/SiO2 | 7.5 | 34 | — | — | 250 | 18000 | 0.1 | [4] |
| 1/5 | Sn-Cu-Na/SiO2 | 0.95 | 58 | — | — | 250 | 20 000 | 0.1 | [50] |
| 2/1 | Rhombic dodecahedra1 Cu2O | 0.8 | 13 | 57 | 5.26 | 250 | 15 000 | 0.1 | [35] |
| 1/2 | 2Cu/5Ru/NaC1/c-SiO2 | 9.4 | 35.6 | — | — | 300 | 20 000 | 0.1 | [62] |
| 1/2 | RuO2-CuO-NaCl/SiO2 | 10 | 40 | — | — | 250 | 30 000 | 0.1 | [51] |
| 1/2 | 2%Cu-5%Ru-1.75 %NaCl/c-SiO2 | 9.6 | 36 | — | — | 300 | 20 000 | 0.1 | [63] |
| 1/5 | Sb2O3-CuO-NaCl/SiO2 | 0.66 | 43 | — | — | 250 | 20 000 | 0.1 | [64] |
| 2/1 | Ag8Cu1/Cs2O-α-Al2O3 | 5.5 | 48.5 | — | — | 160 | 2 000 | 0.1 | [25] |
| 10/31 | RuO2-CuO-Cs2O-TiO2/SiO2 | [0.1 | 7.1 | 52 000 | — | 272 | 848 | 0.1 | [52] |
| 1/2 | RuO2-CuO-NaCl-TeO2-MnOx/SiO2 | 14.55 | 23.1 | 21 690 | — | 297 | 848 | 0.1 | [53] |
| 12/1 | RuO2-CuO-NaCl-TeO2-MnOx /SiO2 | 5.9 | 25.3 | 25 980 | — | 297 | 152 727 | 0.1 | [65] |
| 2/1 | Ag-CuCl2/BaCO3 | 1.3 | 71.2 | — | 0.72 | 200 | 3 000 | 0.1 | [66] |
| 1/4 | RuO2-CuO-TeO2/SiO2 | 0.35 | 47 | 4400 | — | 250 | 152 727 | 0.1 | [58] |
| 2/1 | Ag-Cu-Cl/BaCO3 | 1.2 | 83.7 | — | — | 200 | 3 000 | 0.1 | [57] |
为了方便研究者更快速地进入该领域,下面列举了近十几年来的10篇代表性工作:
(1) 2005年,Lambert等[13]第一次用1% Cu/SiO2作为催化剂进行DEP反应得到了53%的PO催化选择性,首次提出使用Cu基催化剂进行DEP反应;
(2) 2007年,Torres等[14]计算了Cu(111)和Ag(111)表面DEP反应机理,并发现Cu对生成PO更具选择性;
(3) 2008年,Wang等[67]用不同的碱金属和碱土金属对Cu基催化剂进行修饰,发现K+离子具有很好的改性效果;
(4) 2008年,Zhu等[27]确认了Cu+是K+-5% CuOx-SiO2进行DEP反应的活性相,首次详细报道了Cu基催化剂在DEP过程中的状态变化;
(5) 2009年,Su等[26]使用红外光谱考察发现Cu基催化剂对丙烯的吸附能力按Cu0 > Cu+ > Cu2+递减,说明了Cu0和Cu+是活性相,展示了谱学手段对探征反应机理的重要性;
(6) 2011年,Seubsai等[49]在微型反应器中,使用RuO2-CuOx-NaCl催化剂得到40%~50%的PO选择性和10%~20%的丙烯转化率,这个体系是目前报道最高的收率数据;
(7) 2013年,Marimuthu等[24]利用LSPR效应将表层的Cu2O还原成Cu0,较大地提高了PO选择性,提出利用光来调节Cu基催化剂的表面状态;
(8) 2014年,Hua等[35]利用Cu2O纳米模型催化剂研究DEP反应,证实了Cu2O(110)面比Cu2O(100)和Cu2O(111)面更具PO选择性;
(9) 2018年,Song等[36]首次利用DFT计算综合考虑了不同吸附态氧物种对DEP反应的影响,提出O2-可能是生成PO的活性氧物种;
(10) 2018年,Teran等[55]使用现场原位X射线吸收近边结构分析CuOx/SiO2在DEP反应中的Cu价态和修饰剂Na和Ca的影响,展示现场原位技术的重要性。
然而,Cu基催化剂仍然面临着许多问题和挑战,我们认为以下3个领域是未来发展的重要方向:
(1) 构建接近真实条件的模型体系进行DEP反应。Hua等[41]提出利用明确表面结构的Cu2O微纳米晶研究DEP反应的构效关系,然而他们使用的催化剂颗粒尺寸较大、无载体和助剂等修饰,与实际的工业催化剂存在很大的差距。如何构建更加接近工业催化的模型催化体系对于研究催化作用分子机制具有重大意义。
(2) 利用现场原位谱学手段对Cu基催化剂的催化过程进行时实检测。Li等[26, 68]利用原位红外表征了丙烯、O2和PO在Cu基催化剂上的吸附行为,Teran等[55]使用现场原位X射线吸收近边结构分析了DEP反应中Cu的价态变化。除此之外,原位XRD可以分析物相变化,原位拉曼可以表征O2的吸附态,原位XPS可以分析各元素的价态和成分变化,等等。如何在反应条件、反应气氛下实时跟踪Cu基催化剂、载体和助剂的状态,对于深入理解DEP催化性能变化的原因具有非常重要的意义。
(3) 目前DEP反应的研究大部分都是热反应,PO的收率通常低于1%。如何引入光和电等外场,改变Cu基催化剂的表面价态或促进某些关键步骤的转化从而提高DEP的收率是一个重要挑战。Marimuthu等[24]的工作已展示了光对表面氧化态的调控,另外光照也会改变O2在一些金属[69]和金属氧化物[70]上的吸附和转化。电化学调控DEP反应目前还未见报道,但是利用电场可以有效调控催化剂表面氧物种的类型[71],可能可以有效抑制DEP的副反应。
总的来说,Cu基催化剂进行DEP反应具有很好的工业应用前景,但是目前的研究还远未达到工业生产的要求。通过构建接近真实条件的模型体系,并运用现场原位谱学技术时实检测Cu基催化剂的实际情况,并偶合光/电等外场,可以更深层次地揭示催化构效关系并认识其调控机制,为高性能工业催化剂的开发提供新思路。
Ji J, Lu Z, Lei Y, et al.. Catalysts, 2018, 8(10):421 doi: 10.3390/catal8100421
Khatib S J, Oyama S T.. Catal. Rev., 2015, 57(3):306-344 doi: 10.1080/01614940.2015.1041849
Xi Z, Zhou N, Sun Y, et al.. Science, 2001, 292(5519):1139-1141 doi: 10.1126/science.292.5519.1139
He J, Zhai Q, Zhang Q, et al.. J. Catal., 2013, 299:53-66 doi: 10.1016/j.jcat.2012.11.032
Chu H, Yang L, Zhang Q, et al.. J. Catal., 2006, 241(1):225-228 doi: 10.1016/j.jcat.2006.04.028
Nijhuis T A, Makkee M, Moulijn J A, et al.. Ind. Eng. Chem. Res., 2006, 45(10):3447-3459 doi: 10.1021/ie0513090
Min B K, Friend C M.. Chem. Rev., 2007, 107(6):2709-2724 doi: 10.1021/cr050954d
Cowell J J, Santra A K, Lindsay R, et al.. Surf. Sci., 1999, 437(1/2):1-8
Cowell J J, Santra A K, Lambert R M.. J. Am. Chem. Soc., 2000, 122(10):2381-2382 doi: 10.1021/ja994125j
Santra A K, Cowell J J, Lambert R M.. Catal. Lett., 2000, 67(2/3/4):87-91 https://www.researchgate.net/publication/226161426_Ultra-selective_epoxidation_of_styrene_on_pure_Cu111_and_the_effects_of_Cs_promotion
Cropley R L, Williams F J, Vaughan O P H, et al.. Surf. Sci., 2005, 578(1/2/3):L85-L88 https://www.ncbi.nlm.nih.gov/pubmed/15834477
Lambert R M, Williams F J, Cropley R L, et al.. J. Mol. Catal. A-Chem., 2005, 228(1/2):27-33
Vaughan O P H, Kyriakou G, Macleod N, et al.. J. Catal., 2005, 236(2):401-404 doi: 10.1016/j.jcat.2005.10.019
Torres D, Lopez N, Illas F, et al.. Angew. Chem. In. Ed., 2007, 119(12):2101-2104 doi: 10.1002/ange.200603803
王野, 朱文明, 张庆红.催化学报, 2008, 29(9):857-865 doi: 10.3321/j.issn:0253-9837.2008.09.006WANG Ye, ZHU Wen-Ming, ZHANG Qing-Hong.. Chin. J. Catal., 2008, 29(9):857-865 doi: 10.3321/j.issn:0253-9837.2008.09.006
Huang J, Haruta M.. Res. Chem. Intermed., 2012, 38(1):1-24 https://www.researchgate.net/publication/257658553_Gas-phase_propene_epoxidation_over_coinage_metal_catalysts
庞义军, 陈晓晖, 许承志, 等.化学进展, 2014, 26(8):1307-1316 http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201408004.htmPANG Yi-Jun, CHEN Xiao-Hui, XU Cheng -Zhi, et al.. Prog. Chem., 2014, 26(8):1307-1316 http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201408004.htm
Zemichael F W, Palermo A, Tikhov M S, et al.. Catal. Lett., 2002, 80(3/4):93-98 doi: 10.1023/A:1015484121891
Lu J, Bravo-Suárez J J, Haruta M, et al.. Appl. Catal. A-Gen., 2006, 302(2):283-295 doi: 10.1016/j.apcata.2006.01.023
Lei Y, Mehmood F, Lee S, et al.. Science, 2010, 328(5975):224-228 doi: 10.1126/science.1185200
Huang J, Akita T, Faye J, et al.. Angew. Chem. In. Ed., 2009, 48(42):7862-7866 doi: 10.1002/anie.200903011
Dai Y, Chen Z, Guo Y, et al.. Phys. Chem. Chem. Phys., 2017, 19(36):25129-25139 doi: 10.1039/C7CP02892J
Greiner M T, Jones T E, Johnson B E, et al.. Phys. Chem. Chem. Phys., 2015, 17(38):25073-25089 doi: 10.1039/C5CP03722K
Marimuthu A, Zhang J, Linic S.. Science, 2013, 339(6127):1590-1593 doi: 10.1126/science.1231631
Zheng X, Guo Y L, Guo Y, et al.. Rare Metals, 2015, 34(7):477-490 doi: 10.1007/s12598-015-0500-y
Su W G, Wang S G, Ying P L, et al.. J. Catal., 2009, 268(1):165-174 doi: 10.1016/j.jcat.2009.09.017
Zhu W, Zhang Q, Wang Y.. J. Phys. Chem. C, 2008, 112(20):7731-7734 doi: 10.1021/jp800927y
Düzenli D, Atmaca D O, Gezer M G, et al.. Appl. Surf. Sci., 2015, 355:660-666 doi: 10.1016/j.apsusc.2015.07.155
Seubsai A, Zohour B, Noon D, et al.. ChemCatchem, 2014, 6(5):1215-1219 https://www.researchgate.net/publication/261331185_Key_Mechanistic_Insight_into_the_Direct_Gas-Phase_Epoxidation_of_Propylene_by_the_RuO2-CuO-NaClSiO2_Catalyst
Huang W, Sun G, Cao T.. Chem. Soc. Rev., 2017, 46(7):1977-2000 doi: 10.1039/C6CS00828C
Zhang J, Li H, Kuang Q, et al.. Acc. Chem. Res., 2018, 51(11):2880-2887 doi: 10.1021/acs.accounts.8b00344
Zhang J, Kuang Q, Jiang Y, et al.. Nano Today, 2016, 11(5):661-677 doi: 10.1016/j.nantod.2016.08.012
Kuang Q, Wang X, Jiang, Z, et al.. Acc. Chem. Res., 2014, 47(2):308-318 doi: 10.1021/ar400092x
Wang X, Liu C, Zheng B, et al.. J. Mater. Chem. A, 2013, 1(2):282-287 doi: 10.1039/C2TA00241H
Hua Q, Cao T, Gu X K, et al.. Angew. Chem. In. Ed., 2014, 53(19):4856-4861 doi: 10.1002/anie.201402374
Song Y Y, Wang G C.. J. Phys. Chem. C, 2018, 122(37):21500-21513 doi: 10.1021/acs.jpcc.8b07044
Wang Q, Kuang Q, Wang K, et al.. RSC Adv., 2015, 5(75):61421-61425 doi: 10.1039/C5RA08988C
WANG Qiu-Xiang(王秋祥).. Thesis for the Doctorate of Xiamen University(厦门大学博士论文)., 2019.
Du G F, Pei J, Jiang Z Y, et al.. Sci. Bull., 2018, 63(14):892-899 doi: 10.1016/j.scib.2018.05.035
Chen Q, Du G, Dong Y, et al.. Sci. Bull., 2017, 62(20):1359-1364 doi: 10.1016/j.scib.2017.09.008
Chen Q, Cao Z, Du G, et al.. Nano Energy, 2017, 39:582-589 doi: 10.1016/j.nanoen.2017.07.041
Chen Q, Yang Y, Cao Z, et al.. Angew. Chem. Int. Ed., 2016, 55(31):9021-9025 doi: 10.1002/anie.201602592
Zheng X, Zhang Q, Guo Y, et al.. J. Mol. Catal. A-Chem., 2012, 357:106-111 doi: 10.1016/j.molcata.2012.01.027
Chimento R J, Medina F, Fierro J L G, et al.. J. Mol. Catal. A-Chem., 2007, 274(1/2):159-168
Kizilkaya A C, Senkan S, Onal I.. J. Mol. Catal. A-Chem., 2010, 330(1/2):107-111 https://www.sciencedirect.com/science/article/abs/pii/S138111691000302X?via%3Dihub
Bracey C L, Carley A F, Edwards J K, et al.. Catal. Sci. Technol., 2011, 1(1):76-85 doi: 10.1039/c0cy00003e
Duzenli D, Seker E, Senkan S, et al.. Catal. Lett., 2012, 142(10):1234-1243 doi: 10.1007/s10562-012-0867-4
Yang L, He J, Zhang Q, et al.. J. Catal., 2010, 276(1):76-84 doi: 10.1016/j.jcat.2010.09.002
Seubsai A, Kahn M, Senkan S.. ChemCatChem, 2011, 3(1):174-179 doi: 10.1002/cctc.201000248
Miller A, Zohour B, Seubsai A, et al.. Ind. Eng. Chem. Res., 2013, 52(28):9551-9555 doi: 10.1021/ie4004688
Zohour B, Noon D, Seubsai A, et al.. Ind. Eng. Chem. Res., 2014, 53(14):6243-6248 doi: 10.1021/ie402416s
Chukeaw T, Seubsai A, Phon-in P, et al.. RSC Adv., 2016, 6(61):56116-56126 doi: 10.1039/C6RA12559J
Phon-in P, Seubsai A, Chukeaw T, et al.. Catal. Commun., 2016, 86:143-147 doi: 10.1016/j.catcom.2016.08.035
HušM, Hellman A.. J. Catal., 2018, 363:18-25 doi: 10.1016/j.jcat.2018.04.008
Teržan J, Djinovi P, Zavašnik J, et al.. Appl. Catal. B-Environ., 2018, 237:214-227 doi: 10.1016/j.apcatb.2018.05.092
Seubsai A, Senkan S.. ChemCatchem, 2011, 3(11):1751-1754 doi: 10.1002/cctc.201100178
Zhang Q, Guo Y, Zhan W, et al.. Chin. J. Catal., 2017, 38(1):65-72
Seubsai A, Uppala C, Tiencharoenwong P, et al.. Catal. Lett., 2017, 148(2):586-600 https://www.researchgate.net/publication/321673210_High_Stability_of_Ruthenium-Copper-Based_Catalysts_for_Epoxidation_of_Propylene
Lu J, Luo M, Lei H, et al.. J. Catal., 2002, 211(2):552-555 doi: 10.1016/S0021-9517(02)93753-X
Luo M, Lu J, Li C.. Catal. Lett., 2003, 86(1/2/3):43-49 https://www.sciencedirect.com/science/article/abs/pii/S0926860X02000625
Long W, Zhai Q, He J, et al.. ChemPlusChem, 2012, 77(1):27-30 doi: 10.1002/cplu.201100050
Kalyoncu S, Düzenli D, Onal I, et al.. Catal. Lett., 2014, 145(2):596-605
Kalyoncu S, Düzenli D, Onal I, et al.. Catal. Commun., 2015, 61:16-20 doi: 10.1016/j.catcom.2014.12.002
Seubsai A, Noon D, Chukeaw T, et al.. Ind. Eng. Chem. Res., 2015, 32:292-297 doi: 10.1016/j.jiec.2015.08.026
Seubsai A, Phon-in P, Chukeaw T, et al.. Ind. Eng. Chem. Res., 2016, 56(1):100-110
Zhang Q, Chai G, Guo Y, et al.. J. Mol. Catal. A-Chem., 2016, 424:65-76 doi: 10.1016/j.molcata.2016.08.019
Wang Y, Chu H, Zhu W, et al.. Catal. Today, 2008, 131(1/2/3/4):496-504
鲁继青, 吴自力, 罗孟飞, 等.催化学报, 2004, 25(11):855-861 doi: 10.3321/j.issn:0253-9837.2004.11.004LU Ji-Qin, WU Zi-Li, LUO Meng-Fei, et al.. Chin. J. Catal., 2004, 25(11):855-861 doi: 10.3321/j.issn:0253-9837.2004.11.004
Vankayala R, Sagadevan A, Vijayaraghavan P, et al.. Angew. Chem. In. Ed., 2011, 50(45):10640-10644 doi: 10.1002/anie.201105236
Jing X L, Chen Q C, He C, et al.. Phys. Chem. Chem. Phys., 2012, 14(19):6898-6904 doi: 10.1039/c2cp40086c
Seh Z W, Kibsgaard J, Dickens C F, et al.. Science, 2017, 355(6321):eaad4998 doi: 10.1126/science.aad4998

图 2
(A) 币族金属催化剂的DEP反应性能(Au、Ag和Cu分别用正方形、三角形和圆表示), 黑色区域为工业化目标[16];
(B)在Cu(111)、Ag(111)和Au(111)表面从OMMP生成PO和丙酮(AC)的反应能垒[22]
Figure 2 (A) Catalytic performance for DEP over coinage metal catalysts (Au, Ag and Cu are represented as squares, triangles and circles respectively), Industrialized targets are marked as black zone[16]; (B) Reaction barriers for PO and acetone (AC) formation on the Cu(111), Ag(111) and Au(111) surfaces from OMMP[22]


图 4 催化剂Cs+-5% CuOx/SiO2经过(A)在773 K用H2预处理和(B) H2预处理后在573 K用N2O预处理后进行DEP反应的原位XRD图,反应条件是:温度为523 K, 丙烯和O2的分压为50.7 kPa, 总流速为60 mL·min-1; (C, D)在丙烯和O2的持续脉冲输入时Cs+-5% CuOx-SiO2催化剂的DEP反应催化性能[4]
Figure 4 In situ XRD patterns of the Cs+-5% CuOx/SiO2 catalysts pretreated with (A) H2 at 773 K and (B) H2 and followed by N2O at 573 K under reaction conditions (T=523 K, PC3H6=PO2=50.7 kPa, F=60 mL·min-1) for different times; (C, D) Catalytic performance of the Cs+-5% CuOx-SiO2 catalyst after different pretreatments under successive(C3H6+O2) pulses[4]


图 6 (A) Cu2O(111)、(100)和(110)表面及(B) C3H6吸附物种结构:红色、灰色、白色、粉红色和绿色球分别代表O、C、H、配位饱和的Cu和配位不饱和的Cu[35]
Figure 6 (A) Optimized structures of Cu2O(111), (100), and (110) surfaces and (B) relatively adsorbed the most stable C3H6 (a)species: Red, gray, white, pink, and green balls represent O, C, H, coordinatively saturated Cu, and coordinatively unsaturated Cu, respectively[35]



图 9 (A) 修饰了不同碱金属或碱土金属的1% CuOx/SBA-15催化剂的DEP反应催化性能; (B)催化剂1% CuOx/SBA-15和K+-1% CuOx/SBA-15进行DEP反应的阿伦尼乌斯线图[27]
Figure 9 (A) Catalytic performance of 1% CuOx/SBA-15 modified with different alkali metal or alkaline earth metal for DEP reaction; (B) Arrhenius plots of 1% CuOx/SBA-15 and K+-1% CuOx/SBA-15 catalysts for DEP reaction[27]
表 1 近年来Cu基催化剂的DEP反应催化性能
Table 1. Catalytic performances of DEP over different Cu-based catalysts in recent years
| nC3H6/nO2 | Catalyst | Conv./% | Se1ect./% | PO formation rate / (μmo1·g-1·h-1) | T0F/ h-1 | Temp./℃ | Space ve1ocity / h-1 | Pressure/MPa | Ref. |
| 10/1 | VCe0.2Cu0.8-NaC1 | 0.19 | 43.4 | 162.5 | — | 250 | — | 0.1 | [59] |
| 1/2 | Ag-CuC1 | 2.33 | 33.6 | — | — | 350 | 18 000 | 0.1 | [60] |
| 1/1 | 1%Cu/SiO2 | 0.25 | 53 | — | 0.09 | 225 | 30 000 | 0.1 | [13] |
| 1/1 | K+-1% CuOx/SBA-15 | 0.7 | 41 | 2 100 | 17.5 | 350 | 18 000 | 0.1 | [5] |
| 6/5 | K+-1% CuOx/SBA-15 | 2.2 | 24 | 2 200 | 18 | 350 | 18 000 | 0.1 | [27] |
| 1/1 | K+-5% CuOx-SiO2 | 2.0 | 25 | 2 000 | — | 275 | 18 000 | 0.1 | [27] |
| 1/1 | KAc-Cu/SiO2 | 1.3 | 61 | — | — | 275 | 30 000 | 0.1 | [26] |
| 2/1 | VOx-Cu | 2.7 | 16 | 230 | — | 230 | 15 000 | 0.1 | [48] |
| 22/9 | CuAuDSiO2 | 0.13 | 70 | — | — | 300 | 22 500 | 0.1 | [46] |
| 4/1 | RuO2-Cu0-NaC1/Si02 | 10 | 50 | — | — | 250 | 20 000 | 0.1 | [49] |
| 1/2 | 3%Cu-2.25%K/m-SiO2 | 2.90 | 20.49 | 1 549 | — | 350 | 20 000 | 0.1 | [47] |
| 1/39.5 | RuOx-CuOx/SiO2 | 13 | 26 | — | — | 225 | 18 000 | 0.1 | [61] |
| 2/1 | Ag-CuDBaCO3 | 3.6 | 55.1 | — | — | 200 | 2 000 | 0.1 | [43] |
| 1/39.5 | Cs+-5%CuOx/SiO2 | 7.5 | 34 | — | — | 250 | 18000 | 0.1 | [4] |
| 1/5 | Sn-Cu-Na/SiO2 | 0.95 | 58 | — | — | 250 | 20 000 | 0.1 | [50] |
| 2/1 | Rhombic dodecahedra1 Cu2O | 0.8 | 13 | 57 | 5.26 | 250 | 15 000 | 0.1 | [35] |
| 1/2 | 2Cu/5Ru/NaC1/c-SiO2 | 9.4 | 35.6 | — | — | 300 | 20 000 | 0.1 | [62] |
| 1/2 | RuO2-CuO-NaCl/SiO2 | 10 | 40 | — | — | 250 | 30 000 | 0.1 | [51] |
| 1/2 | 2%Cu-5%Ru-1.75 %NaCl/c-SiO2 | 9.6 | 36 | — | — | 300 | 20 000 | 0.1 | [63] |
| 1/5 | Sb2O3-CuO-NaCl/SiO2 | 0.66 | 43 | — | — | 250 | 20 000 | 0.1 | [64] |
| 2/1 | Ag8Cu1/Cs2O-α-Al2O3 | 5.5 | 48.5 | — | — | 160 | 2 000 | 0.1 | [25] |
| 10/31 | RuO2-CuO-Cs2O-TiO2/SiO2 | [0.1 | 7.1 | 52 000 | — | 272 | 848 | 0.1 | [52] |
| 1/2 | RuO2-CuO-NaCl-TeO2-MnOx/SiO2 | 14.55 | 23.1 | 21 690 | — | 297 | 848 | 0.1 | [53] |
| 12/1 | RuO2-CuO-NaCl-TeO2-MnOx /SiO2 | 5.9 | 25.3 | 25 980 | — | 297 | 152 727 | 0.1 | [65] |
| 2/1 | Ag-CuCl2/BaCO3 | 1.3 | 71.2 | — | 0.72 | 200 | 3 000 | 0.1 | [66] |
| 1/4 | RuO2-CuO-TeO2/SiO2 | 0.35 | 47 | 4400 | — | 250 | 152 727 | 0.1 | [58] |
| 2/1 | Ag-Cu-Cl/BaCO3 | 1.2 | 83.7 | — | — | 200 | 3 000 | 0.1 | [57] |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: