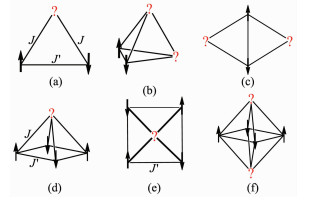
图 1.
在一些特定结构中的自旋耦合竞争与阻挫
Figure 1.
Spin-coupling competition and frustration in some specific structures

二十世纪六七十年代,Toulouse[1]和Villain[2]在研究自旋玻璃时提出阻挫(frustration)这一概念,陈韫春等[3]对其中文描述为:“阻挫是自然界普遍存在的一种现象,它是指体系内部各单元的能量因相互竞争而无法同时满足每个个体能量都最低的一种现象。其结果是,任何一方都无法取得支配地位的优势,而正是这些‘竞争中的失败者’影响整个系统的性质”。自旋阻挫体系建立在几何阻挫的基础上,是指磁耦合作用相互竞争的体系[4]。如图 1所示,在一个三角形体系中,如果2个自旋间的耦合J′占主导性地位而且为反铁磁性,那么第三个自旋的方向将无法确定,即处于阻挫的状态,此时的基态为简并态,再不能根据我们通常的自旋平行或反平行排列简单地确定,而必须通过J′/J来确定。基于三角形结构可以衍生出的化合物,如具有正四面体、四自旋中心蝴蝶型结构、三角双锥、四方锥、中心有自旋的正方形、八面体等(图 1)结构类型的多核簇合物,自旋间适当的耦合竞争都可能导致自旋阻挫。自旋阻挫一直是磁学方面的难点课题,在分子磁学领域,目前能清楚地解释磁结构相关性的自旋阻挫只达到四方锥的五核体系[5]。显然,以3个自旋中心组成的三角形结构可以形成最简单的自旋阻挫关系,其磁结构相关性易于研究,所以是目前报道最多的体系[6-11],其中[Cu3]是代表性化合物。因Cu(Ⅱ)离子只有一个未成对电子,[Cu3]是最简单的阻挫体系,所以其经常作为结构单元出现在二维和三维的自旋阻挫体系中,以获得更有价值的材料,比如量子自旋液体等。
基于肟的配体具有多种配位位点及非共价作用,被广泛用于合成多核磁性分子[6]。它们既可以通过N原子又可以通过O原子连接金属离子,易于通过M-N-O-M′桥联形成同金属或异金属多核化合物[12-14]。其中典型的例子是通过配体骨架修饰合成的三核[6-11, 15]和四核[16-17]铜肟化合物,多表现为几何阻挫结构,是自旋阻挫研究中很好的候选化合物。
2014年,Shi课题组[18]利用吡嗪肟配体合成了三核铜配合物[Cu3(μ3-OH)(μ-OPz)3(NO3)2(H2O)2]·CH3OH (1)和三核银配合物[Ag3(HOPz)2(NO3)3]n,其中三核铜是一个典型的自旋阻挫体系。2个配合物中,吡嗪肟配体与金属的结合位点不同,如果将铜和银在一个反应中进行自组装,有可能得到一种由配合物1和银化合物相互铰链的全新的自旋阻挫体系,和配合物1的磁性进行对比,可以观察不同分子间弱作用对自旋阻挫性质的影响。基于这种构想,我们制备了由银离子连接配合物1形成的一个三维金属有机框架阻挫配合物{[Ag(HOPz)Cu3(μ3-OH)(NO3)3(OPz)2Ag(NO3)]·6H2O}n (2)。在本工作中,我们将对其合成、结构和磁性质进行详细的报导。
所有试剂皆从商业途径获得且无进一步纯化。配体HOPz和配合物1均根据文献方法制备[18]。
在Bruker Tensor 27红外光谱仪上利用溴化钾压片法测试了红外光谱,数据由OPUS软件处理。磁性测量采用Quantum Design MPMS SQUID VSM磁测量系统,磁化率数据经过Pascal常数和样品架背景抗磁校正。使用Bruker SMART Apex Ⅱ CCD单晶仪[19]对晶体结构进行测试,同时由Apex Ⅱ程序收集晶体数据,用SAINT和SADABS程序处理结构数据[20],并由SHELXTL-97程序完成结构解析[21]。其中,非氢原子坐标用差值傅立叶(Fourier)合成法确定,对其原子坐标及其各向异性热参数进行全矩阵最小二乘法精修至收敛;氢原子坐标通过理论加氢法获得并用跨式模型(riding model)进行精修。
CCDC:819086,1;1965094,2。
将HOPz(1.0 mmol,137mg)溶解在10 mL甲醇中。将三水合硝酸铜(1.0 mmol,137mg)溶解在10 mL甲醇中。混合2份溶液,搅拌2 h。加入10 mL硝酸银(0.5 mmol,85mg)的甲醇溶液,过滤静置,蒸发结晶。24 h后出现黑色晶体。
产量:124.5 mg(38.0%)。红外(溴化钾压片, cm-1):3 423,2 395,1 763,1 627,1 590,1 567,1 519,1 468,1 403,1 199,1 168,1 144,1 091,1 041,860,825,757,733,480,418。元素分析按Ag4C36Cu6N26O44H62计算值(%):C 18.20,H 2.63,N 15.33;实测值(%):C 18.16,H 2.75,N 15.13。
单晶X射线衍射分析表明,配合物1和2均结晶在单斜晶系,空间群分别为P21/n和P21/c,晶体学参数见表 1,部分键长键角见表 2。
 下载:
导出CSV
下载:
导出CSV
| Complex | 1 | 2* |
| Formula | C19H27Cu3N11O13 | Ag4C36Cu6N26O44H62 |
| Formula weight | 808.17 | 2 375.78 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c |
| a / nm | 1.373 5(3) | 1.147 5(18) |
| b / nm | 1.492 6(3) | 2.955(5) |
| c / nm | 1.508 8(3) | 1.127 0(19) |
| β / (°) | 101.190(4) | 105.41(3) |
| V / nm3 | 3.034 37 | 3.685(10) |
| Z | 4 | 2 |
| Dc / (g·cm-3) | 1.065 | 1.289 |
| GOF | 1.182 | 1.024 |
| Reflection, restraint, parameter | 6 961, 0, 423 | 6 325, 91, 494 |
| R1 | 0.058 3 | 0.096 2 |
| wR2 | 0.212 2 | 0.294 9 |
| * Chemical formula, relative molecular weight and density of compound 2 are the comprehensive characterization results of single crystal structure, elemental analysis and thermogravimetric analysis. Six water solvent molecules in the cif file are subjected to squeeze treatment. | ||
下载:
导出CSV
| 1 | |||||
| Cu-O10 | 0.195 5(3) | Cu2-O10 | 0.197 2(9) | Cu3-O10 | 0.195 0(7) |
| Cu1-O2 | 0.195 8(8) | Cu2-O1 | 0.192(1) | Cu3-O3 | 0.194(1) |
| Cu1-N3 | 0.199(1) | Cu2-N7 | 0.198(1) | Cu3-N6 | 0.199(1) |
| Cu2-O10-Cu3 | 109.5(2) | Cu1-O10-Cu3 | 109.9(2) | Cu1-O10-Cu2 | 113.3(2) |
| 2 | |||||
| Cu1-O10 | 0.195 1(3) | Cu2-O10 | 0.194 6(5) | Cu3-O10 | 0.195 6(4) |
| Cu1-O3 | 0.193 2(5) | Cu2-O4 | 0.193 4(5) | Cu3-O2 | 0.194 0(4) |
| Cu1-N1 | 0.198 1(5) | Cu2-N5 | 0.197 5(5) | Cu3-N4 | 0.198 7(5) |
| Cu3-O10-Cu2 | 109.4(4) | Cu2-O10-Cu1 | 112.2(4) | Cu1-O10-Cu3 | 111.3(4) |
单晶结构显示,配合物1的结构完全和文献的相同[18]。
配合物2与配合物1具有结构相似的[Cu3]单元,只是键长键角发生了微小的变化。由3个Cu(Ⅱ)离子、1个μ3-OH-、3个去质子μ-OPz-配体、2个NO3-和2个H2O分子组成。如图 2a所示,3个Cu(Ⅱ)离子被1个中心羟基μ3-氧桥联,再通过3个OPz-配体的氧氮桥连,同时OPz-配体上吡嗪基提供1个N配位,形成近似的平面三角形骨架。羟基反侧,1个η-硝酸根轴向桥联2个Cu(Ⅱ)离子;羟基侧,1个水分子和1个硝酸根分别轴向配位这2个Cu(Ⅱ)离子,所以二者是六配位扭曲八面体构型。另有水分子从羟基侧轴向配位第三个Cu(Ⅱ)离子,形成五配位四方锥构型。Cu(Ⅱ)离子周围键长键角见表 2。

All hydrogen atoms and solvent molecules are omitted for clarity; Ellipsoid probability: 70%
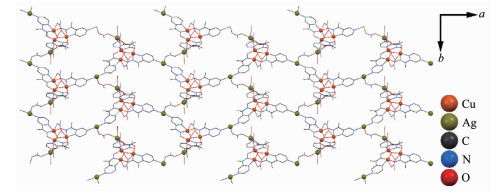
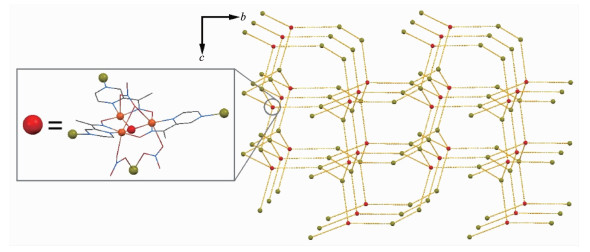
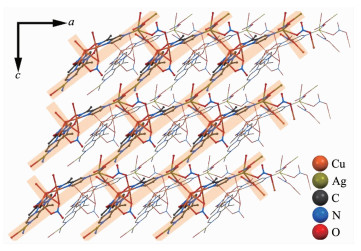
配合物2利用配合物1三角形结构顶点处配体OPz-中剩余的吡嗪N进一步结合Ag?髣离子,将配合物1分子和Ag?髣离子桥联形成三维结构,所以[Cu3]部分的结构中,Cu-μ3O的键长在配合物1中是0.195 5(4)、0.195 2(3)和0.194 4(5) nm,Cu-O-Cu键角为109.5(2)、109.9(2)和113.3(2)°;对应的键长和键角在配合物2中分别是0.195 2(9)、0.197 3(9)和0.194 7(7) nm,109.4(4)°、112.2(4)°和111.3(4)°。其中Ag?髣周围有2种配位构型(图 2b)。三配位平面T字构型部分,配位原子分别来自于同层2个[Cu3]分子的吡嗪N和1个硝酸根(图 3),该硝酸根通过η-桥联到另一个Ag?髣离子,后者又通过错层[Cu3]分子羟基侧的2个η-硝酸根桥联到Cu(Ⅱ)离子,1个水分子配位后形成扭曲三角双锥五配位构型。因为桥联来自不同的层,所以这些连接导致一个三维有机金属框架网状结构(图 4)。在该配合物中,共有2套这样的网状结构,它们相互交错而不相连最终构成配合物2的结构(图 5)。
配合物1和2的变温直流磁化率在1 kOe场强下采用粉末多晶样品进行测试,测量温度范围为1.8~300 K。如图 6和图 7所示,因两配合物中[Cu3]单元相同,所以2条变温磁性曲线形状非常相似。配合物1和2的室温χMT值分别为0.48和0.51 cm3·mol-1·K,远小于3个唯自旋S=1/2(假定g=2.1)的Cu(Ⅱ)理论值1.24 cm3·mol-1·K。这表明分子间有很强的反铁磁性耦合。随着温度降低,χMT值以恒定速率下降至100 K时出现拐点,约为0.4 cm3·mol-1·K,与S=1/2的基态计算值相符。低于拐点的部分,曲线更加陡峭,最低点分别为0.25和0.28 cm3·mol-1·K,说明三核铜内存在强的反铁磁性耦合和反对称交换[22-28]。
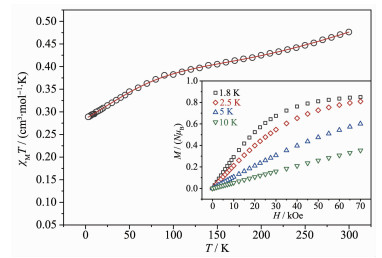
Circle: experimental data, Solid line: the best fit to the experimental data; Inset: Plot of M vs H for complex 1

Circle: experimental data, Solid line: the best fit to the experimental data; Inset: Plot of M vs H for complex 2
为了确定独立交换作用的本质,将铜簇中的3个铜分别占据三角形的一角,则一个分子中的3个磁离子相互间有3个交换作用。每个磁作用包含μ3-氧桥和-OPz-配体的桥连氮氧原子。为了研究此三核铜簇的磁行为,使用各向同性的海森堡-狄拉克-范弗莱克(Heisenberg-Dirac-Van Vleck,HDVV)哈密顿算符进行分析。
|
$ \hat{H}_{\mathrm{iso}}=-2\left[J_{12}\left(\hat{S}_{1} \hat{S}_{2}\right)+J_{23}\left(\hat{S}_{2} \hat{S}_{3}\right)+J_{13}\left(\hat{S}_{1} \hat{S}_{3}\right)\right] $ |
(1) |
其中,Jij是交换积分(磁耦合常数),
因为配合物1和2中的3个铜离子在晶体中是不等价的,需将三核当作不等边三角形,然而不等边三角形中存在3个耦合常数,无法精确地求解,所以将键长键角更接近的2个铜离子视作相等,获得等腰三角形。即
|
$ J_{12}=J_{13}=J^{\prime} $ |
(2) |
则
|
$ \left.\hat{H}_{\mathrm{iso}}=-2 J^{\prime}\left[\left(\hat{S}_{1} \hat{S}_{2}\right)+\left(\hat{S}_{1} \hat{S}_{3}\right)\right]-2 J_{23}\left(\hat{S}_{2} \hat{S}_{3}\right)\right] $ |
(3) |
定义
|
$ \hat{H}_{\mathrm{iso}}=-J^{\prime}\left[\hat{S}_{\mathrm{T}}\left(\hat{S}_{\mathrm{T}^{+}} 1\right)-\hat{S}^{\prime}(\hat{S}+1)\right]-J_{23}\left(\hat{S}\left(\hat{S}^{\prime}+1\right)-2 \hat{S}_{i}\left(\hat{S}_{i}+1\right)\right] $ |
|
$ \begin{aligned} E\left(\hat{S}_{\mathrm{T}}, \hat{S}^{\prime}\right)=&-J^{\prime}\left[\hat{S}_{\mathrm{T}}\left(\hat{S}_{\mathrm{T}}+1\right)-\hat{S}^{\prime}\left(\hat{S}^{\prime}+1\right)-\hat{S}_{i}\left(\hat{S}_{i}+1\right)\right]-\\ & J_{23}\left[\hat{S}^{\prime}\left(\hat{S}^{\prime}+1\right)-2 \hat{S}_{i}\left(\hat{S}_{i}+1\right)\right] \end{aligned} $ |
(4) |
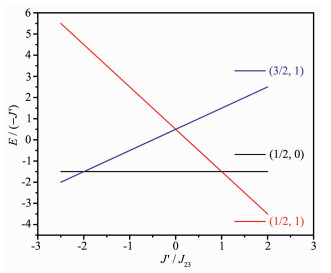
据此,得到三核铜簇的基态能级图。3个铜原子自旋S=1/2,故可能存在2种基态,见图 8。
低温下的磁行为来源于三核铜簇内的反对称交换作用。因为各向同性HDVV模型中,自旋多重态间的旋轨耦合未被考虑,故居里定律的计算值大于反对称交换存在下的磁矩。
反对称交换用哈密顿算符表示如下:
|
$ \hat{H}_{\mathrm{ASE}}=G_{12}\left(\hat{S}_{1} \hat{S}_{2}\right)+G_{23}\left(\hat{S}_{2} \hat{S}_{3}\right)+G_{31}\left(\hat{S}_{3} \hat{S}_{1}\right) $ |
(5) |
其中, Gij是反对称向量参数。
鉴于3个铜离子的结构参数差别很小,为避免拟合时过参数化,令G12=G23=G31=GZ。
图 9展示了三角形三核铜簇在反铁磁作用下的能级裂分。在等边三角形(C3)中,各向同性交换(
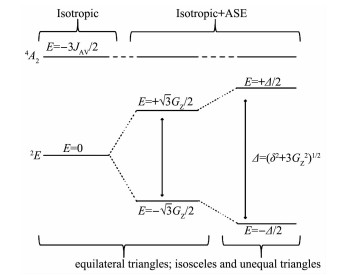
反对称交换作用使得基态能隙增加至
综上条件,加上轴向塞曼哈密顿算符:
|
$ \begin{aligned} \hat{H}_{\mathrm{Zeem}}=& g_{ //} \mu_{\mathrm{B}}\left(\hat{S}_{\mathrm{1z}}+\hat{S}_{2z}+\hat{S}_{\mathrm{3z}}\right) \hat{H}_{z}+\\ & g_{\perp} \mu_{\mathrm{B}}\left[\left(\hat{S}_{{1x}}+\hat{S}_{2 x}+\hat{S}_{3 x}\right) \hat{H}_{x}+\left(\left(\hat{S}_{1 y}+\hat{S}_{2 y}+\hat{S}_{3y}\right) \hat{H}_{y}\right]\right. \end{aligned} $ |
(6) |
其中,μB是玻尔磁子,g∥=g1z=g2z=g3z是平行于z轴的朗德因子,g⊥=g1x=g2x=g3x是垂直于z轴的朗德因子。
获得完整的哈密顿算符如下:
|
$ \hat{H}=\hat{H}_{\mathrm{i} \mathrm{so}}+\hat{H}_{\mathrm{ASE}}+\hat{H}_{\mathrm{Zeem}} $ |
(7) |
由此推出零场下磁化率的数学表达式[28]:
|
$ \chi_{\mathrm{M}}^{//}=\frac{N \mu_{\mathrm{B}}^{2} g_{/ /}^{2}}{4 k T}\left(\frac{A+5 B}{A+B}\right) $ |
(8) |
|
$ \chi_{\mathrm{M}}^{\perp}=\frac{N \mu_{\mathrm{B}}^{2} g_{\perp}^{2}}{4 k T}\left(\frac{\rho^{2} A+5 B+\left(1-\rho^{2}\right) C}{A+B}\right) $ |
(9) |
|
$ \chi_{\mathrm{M}}^{\mathrm{av}}=\frac{\chi_{\mathrm{M}}^{//}+\chi_{\mathrm{M}}^{\perp}}{3} $ |
(10) |
其中,
对配合物1和2的磁化率数据进行拟合,相关参数为Jav,δ,g⊥,g∥和GZ。最佳拟合结果如表 3所示。
下载:
导出CSV
| Complex | -Jav | -J′ | -J23 | δ | GZ | g⊥ | g∥ |
| 1 | 426 | 457 | 363 | 93.9 | 80.2 | 1.83 | 2.00 |
| 2 | 401 | 414 | 376 | 37.9 | 79.7 | 1.85 | 2.00 |
将所得参数代入能级图(图 8),可知2个配合物的基态能级皆为(1/2,1)。
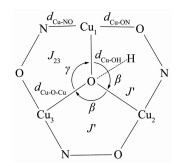
表 4列出了配合物1和2的磁构参数。如图 10,为了描述三角形的晶体几何性质,定义J′对应的桥角β,是数值相近的2个Cu-O-Cu键角的平均值。J23对应的桥角γ,是与其他桥角差别最大的Cu-O-Cu键角值。Jav=(2J′+J23)/3对应的桥角:αav=(2β+γ)/3。
下载:
导出CSV
| J23 / cm-1 | γ / (°) | J′ / cm-1 | β / (°) | Jav / cm-1 | αav / (°) | dCu-ON / nm | dCu-NO / nm | dCu-OH / nm | |
| 1 | -363 | 113.3 | -457 | 109.7 | -426 | 110.9 | 0.193 9 | 0.198 7 | 0.195 8 |
| 2 | -376 | 109.4 | -414 | 111.8 | -401 | 111.0 | 0.193 5 | 0.198 1 | 0.195 1 |
虽然铜离子之间的磁相互作用被中心氧桥和肟基的氮氧桥减弱,但Cu-ON和Cu-NO键长变化与交换耦合无关[7]。肟基作为刚性基团,在三角形中具有确定的磁耦合作用。
因此,对磁性有决定性影响的是Cu-OH键长和Cu-O-Cu桥角。这个角度反映了三核铜簇的平面化程度,即桥角越大,平面化程度越大[6]。通常,配合物的耦合常数J数值与键长和角度成正相关[28]。键角越大,Cu-OH键长越短,磁耦合越强。因为2个配合物结构相近,故两者磁学参数大致接近。
合成了配合物1和2并表征了其结构和磁性。通过包含各向同性和反对称交换的哈密顿算符对其进行拟合,证明了它们的强反铁磁相互作用。验证了三角构型三核铜簇阻挫分子的磁构关系,并为阻挫分子的合成提供了新思路。
Toulouse G. Commun. Phys., 1977, 2:115-119 https://www.researchgate.net/publication/240345794_Theory_of_the_frustration_effect_in_spin_glasses_I
Villain J. J. Phys. Chem. Solids, 1959, 11(3):303-309 https://www.researchgate.net/publication/222908219_La_structure_des_substances_magnetiques
陈韫春, 李丹, 马星桥.物理与工程, 2007, 6:46-48 http://www.cnki.com.cn/Article/CJFDTotal-GKWL200706016.htmCHEN Yun-Chun, LI Dan, MA Xing-Qiao. Physics and Engineering, 2007, 6:46-48 http://www.cnki.com.cn/Article/CJFDTotal-GKWL200706016.htm
Diep H T. Frustrated Spin Systems. Singapore: World Scientific Publishing Co. Pte. Ltd., 2005.
Cao F, Wei R M, Li J, et al. Inorg. Chem., 2016, 55(12):5914 -5923 doi: 10.1021/acs.inorgchem.6b00255
Das L K, Drew M G B, Diaz C, et al. Dalton Trans., 2014, 43 (20):7589-7598 doi: 10.1039/C3DT53361A
Escuer A, Vlahopoulou G, Lloret F, et al. Eur. J. Inorg. Chem., 2014(1):83-92
Maity D, Mukherjee P, Ghosh A, et al. Eur. J. Inorg. Chem., 2010(5):807-813
Sarkar B, Ray M S, Drew M G B, et al. Polyhedron, 2006, 25 (16):3084-3094 doi: 10.1016/j.poly.2006.05.032
Vasylevs'kyy S I, Senchyk G A, Lysenko A B, et al. Inorg. Chem., 2014, 53(7):3642-3654 doi: 10.1021/ic403148f
Zhu T T, Sun W, Huang Y X, et al. J. Mater. Chem. C, 2014, 2(38):8170-8178 doi: 10.1039/C4TC01416B
Chakravorty A. Coord. Chem. Rev., 1974, 13(1):1-46 doi: 10.1016/S0010-8545(00)80250-7
Chaudhuri P. Coord. Chem. Rev., 2003, 243(1/2):143-190
Keeney M E, Osseo-Asare K, Woode K A. Coord. Chem. Rev., 1984, 59:141-201 doi: 10.1016/0010-8545(84)85054-7
Gatteschi D, Caneschi A, Sessoli R. NATO Advanced Science Institutes Series, Series E, Applied Sciences: Vol.206. Laine R M. Ed., Dordrecht: Kluwer Academic, 1992:147-160
Giri S, Maity D, Godsell J F, et al. Inorg. Chim. Acta, 2011, 377(1):99-104
Ruiz R, Lloret F, Julve M, et al. Inorg. Chim. Acta, 1998, 268(2):263-269
Zhang G Y, Ding B, Chai L, et al. Z. Anorg. Allg. Chem., 2014, 640(12/13):2492-2497
Bruker Analytical X-ray Systems, SMART & SAINT Software Reference Manuals, Ver. 6.45, Madison, WI, 2003.
Sheldrick G M. SADABS: Software for Empirical Absorption Correction, Ver. 2.05, University of Gttingen, Germany, 2002.
Sheldrick G M. SHELXL-97: Program for Crystal Structure Refinement, University of Göttingen, Germany, 1997.
Liu X M, de Miranda M P, McInnes E J L, et al. Dalton Trans., 2004(1):59-64 doi: 10.1039/B311980G
Yoon J, Mirica L M, Stack T D P, et al. J. Am. Chem. Soc., 2004, 126(39):12586-12595 doi: 10.1021/ja046380w
Mirica L M, Stack T D P. Inorg. Chem., 2005, 44(7):2131-2133 doi: 10.1021/ic048182b
Afrati T, Dendrinou-Samara C, Raptopoulou C, et al. Dalton Trans., 2007(44):5156-5164 doi: 10.1039/b708767e
Afrati T, Dendrinou-Samara C, Raptopoulou C, et al. Inorg. Chem., 2008, 47(17):7545-7555 doi: 10.1021/ic8003257
Afrati T, Pantazaki A A, Dendrinou-Samara C, et al. Dalton Trans., 2010, 39(3):765-775 doi: 10.1039/B914112J
Ferrer S, Lloret F, Pardo E, et al. Inorg. Chem., 2012, 51(2): 985-1001 doi: 10.1021/ic2020034

图 1 在一些特定结构中的自旋耦合竞争与阻挫
Figure 1 Spin-coupling competition and frustration in some specific structures

图 2 配合物1 (a)和配合物2 (b)的分子结构
Figure 2 Crystal structures of complexes 1 (a) and 2 (b)
All hydrogen atoms and solvent molecules are omitted for clarity; Ellipsoid probability: 70%

图 6 配合物1的χMT形式下的变温直流磁化率曲线
Figure 6 Plot of χMT vs T for complex 1
Circle: experimental data, Solid line: the best fit to the experimental data; Inset: Plot of M vs H for complex 1

图 7 配合物2的χMT形式下的变温直流磁化率曲线
Figure 7 Plot of χMT vs T for complex 2
Circle: experimental data, Solid line: the best fit to the experimental data; Inset: Plot of M vs H for complex 2

图 8 三核铜簇基态能级图
Figure 8 Dependence of ground energy level on the parameter J′/J23 for trinuclear copper cluster

图 9 等边三角形、等腰三角形和不等边三角形在各向同性和反对称交换下的能级图
Figure 9 Energy level diagrams for equilateral triangles, isosceles triangles and unequal triangles under isotropic and antisymmetric exchanges

图 10 肟配体三核铜簇的键长键角命名
Figure 10 Abbreviations of structural and coupling parameters of oxime ligand trinuclear copper cluster
表 1 配合物1和2的晶体数据和结构精修参数
Table 1. Crystal data and structural refinement parameters for complexes 1 and 2
| Complex | 1 | 2* |
| Formula | C19H27Cu3N11O13 | Ag4C36Cu6N26O44H62 |
| Formula weight | 808.17 | 2 375.78 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/n | P21/c |
| a / nm | 1.373 5(3) | 1.147 5(18) |
| b / nm | 1.492 6(3) | 2.955(5) |
| c / nm | 1.508 8(3) | 1.127 0(19) |
| β / (°) | 101.190(4) | 105.41(3) |
| V / nm3 | 3.034 37 | 3.685(10) |
| Z | 4 | 2 |
| Dc / (g·cm-3) | 1.065 | 1.289 |
| GOF | 1.182 | 1.024 |
| Reflection, restraint, parameter | 6 961, 0, 423 | 6 325, 91, 494 |
| R1 | 0.058 3 | 0.096 2 |
| wR2 | 0.212 2 | 0.294 9 |
| * Chemical formula, relative molecular weight and density of compound 2 are the comprehensive characterization results of single crystal structure, elemental analysis and thermogravimetric analysis. Six water solvent molecules in the cif file are subjected to squeeze treatment. | ||
 下载: 导出CSV
下载: 导出CSV
表 2 配合物1和2的部分键长(nm)和键角(°)
Table 2. Selected bond lengths (nm) and bond angles (°) for complexes 1 and 2
| 1 | |||||
| Cu-O10 | 0.195 5(3) | Cu2-O10 | 0.197 2(9) | Cu3-O10 | 0.195 0(7) |
| Cu1-O2 | 0.195 8(8) | Cu2-O1 | 0.192(1) | Cu3-O3 | 0.194(1) |
| Cu1-N3 | 0.199(1) | Cu2-N7 | 0.198(1) | Cu3-N6 | 0.199(1) |
| Cu2-O10-Cu3 | 109.5(2) | Cu1-O10-Cu3 | 109.9(2) | Cu1-O10-Cu2 | 113.3(2) |
| 2 | |||||
| Cu1-O10 | 0.195 1(3) | Cu2-O10 | 0.194 6(5) | Cu3-O10 | 0.195 6(4) |
| Cu1-O3 | 0.193 2(5) | Cu2-O4 | 0.193 4(5) | Cu3-O2 | 0.194 0(4) |
| Cu1-N1 | 0.198 1(5) | Cu2-N5 | 0.197 5(5) | Cu3-N4 | 0.198 7(5) |
| Cu3-O10-Cu2 | 109.4(4) | Cu2-O10-Cu1 | 112.2(4) | Cu1-O10-Cu3 | 111.3(4) |
下载: 导出CSV
表 3 配合物1和2的最佳拟合磁性参数
Table 3.
Best-fit magnetic parameters for complexes 1 and 2
| Complex | -Jav | -J′ | -J23 | δ | GZ | g⊥ | g∥ |
| 1 | 426 | 457 | 363 | 93.9 | 80.2 | 1.83 | 2.00 |
| 2 | 401 | 414 | 376 | 37.9 | 79.7 | 1.85 | 2.00 |
下载: 导出CSV
表 4 配合物1和2的磁构参数
Table 4. Magnetostructural data for complexes 1 and 2
| J23 / cm-1 | γ / (°) | J′ / cm-1 | β / (°) | Jav / cm-1 | αav / (°) | dCu-ON / nm | dCu-NO / nm | dCu-OH / nm | |
| 1 | -363 | 113.3 | -457 | 109.7 | -426 | 110.9 | 0.193 9 | 0.198 7 | 0.195 8 |
| 2 | -376 | 109.4 | -414 | 111.8 | -401 | 111.0 | 0.193 5 | 0.198 1 | 0.195 1 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: