
图 1.
SCN和BM-SCN-x样品的XRD图
Figure 1.
XRD patterns of SCN and BM-SCN-x samples

随着工业化和城市化的持续发展,人类面临的环境污染问题日益严重。半导体光催化技术能直接降解和矿化有机污染物,具有能耗低、反应快和无二次污染等优势,是从根本上解决这些问题的理想途径之一[1]。近年来,一种不含金属元素的聚合物光催化剂石墨相氮化碳(g-C3N4),在光催化领域引起了研究的热潮。作为可见光驱动的有机半导体光催化剂,g-C3N4具有高化学稳定性、低成本、无污染以及适当带隙能量等[2]许多独特的性质,在光催化领域显示出了巨大的潜力[3-5]。但单纯的g-C3N4存在表面缺陷较多,比表面积低,光生电子-空穴复合率高的问题,导致光催化性能较差[6]。为此,人们开发了许多方法以提高g-C3N4的光催化性能,如形貌的调控[7]、金属或非金属元素的掺杂[8]以及异质结的构建[9]。
其中,痕量(< 1.0%(w/w))的非原位和原位硫掺杂是改善g-C3N4光催化性能的常见手段,它可以降低g-C3N4的带隙,显著增强其在可见光区域的吸收强度,从而提高光催化活性[10-12]。硫脲被认为是构建原位硫掺杂g-C3N4的有效前驱体[13-15]。Hong等[14]使用SiO2纳米颗粒作为硬模板,由单一前驱体硫脲首次合成了原位硫掺杂的介孔g-C3N4。研究表明,硫取代了g-C3N4中的碳原子并降低了价带,其光解水制氢的速率是g-C3N4的30.0倍。Lu等[15]用硫取代g-C3N4中的晶格氮,改变了g-C3N4的电子结构,使其具有较窄的带隙和良好的载流子迁移率,该材料在可见光下将可溶性铀?髩还原成不溶性铀(Ⅳ)的催化反应速率是g-C3N4的1.8倍。虽然不少研究已证明原位硫掺杂g-C3N4比单纯的g-C3N4具有更窄的带隙宽度和更强的吸光能力,但是其比表面积仍然不高,表面缺陷也没有减少,导致其光催化活性受到限制。
球磨法是近年来快速发展的材料制备新方法,它利用机械能来诱发化学反应,导致材料的结构和性能发生变化。由于球磨反应在固态下进行,不需要溶剂再生和处理,所以整个球磨过程绿色环保,简单易行,已被证明是改善光催化剂可见光活性的有效方法[16-19]。Zhu等[18]使用球磨法一步合成了剥离的g-C3N4纳米片,与块状g-C3N4相比,其对罗丹明B的光催化降解速率提高了3.0倍以上,这归因于形貌和比表面积对光催化性能的关键作用。Kim等[19]发现在球磨48 h后g-C3N4的聚合度增加,表面缺陷明显减少,制氢速率比未球磨的g-C3N4提高了4.5倍。然而,至今还没有通过球磨法对硫掺杂g-C3N4进行改性来降解水中有机污染物的报道。
本研究通过高温煅烧硫脲以制备原位硫掺杂g-C3N4,按照不同物料溶剂比进行球磨改性,采用多种表征技术对催化剂结构进行分析,并在可见光照射下探讨球磨干湿环境对其光催化降解亚甲基蓝(MB)的反应活性的影响。实验结果表明,湿式球磨改性后的硫掺杂g-C3N4比未球磨和干式球磨后的样品具有更强的光催化降解活性。
硫脲和MB购自阿拉丁生化科技股份有限公司。药品均为分析纯,使用过程中不需要进一步提纯。实验用水为去离子水。
硫掺杂g-C3N4的合成:通过在马弗炉中煅烧硫脲以合成硫掺杂g-C3N4[12]。称取10.0 g硫脲放入带盖坩埚中,在马弗炉中以5 ℃·min-1的升温速率加热至520 ℃并保持2 h,然后冷却至室温。将获得的黄棕色产物研磨成细粉,标记为SCN。
球磨改性硫掺杂g-C3N4的合成:先将氧化锆球和0.5 g SCN以30:1的球料质量比加入到容积为120 mL的氧化锆罐中,再分别将0、1.0、2.0 mL的去离子水用作分散剂混合均匀。采用立式行星球磨机(YXQM-2L,长沙米淇仪器设备有限公司)将样品以600 r·min-1的速度研磨8 h。研磨后得到的粉末在80℃下干燥12 h,按照物料溶剂比分别将样品标记为BM-SCN-0、BM-SCN-1、BM-SCN-2(BM-SCN-x,即x=0,1,2)。
采用全自动X射线衍射仪(D8 Advance,德国布鲁克AXS公司)进行物相分析,靶源为Cu Kα射线(λ=0.154 056 nm),测试电压为40.0 kV,电流为40.0 mA,扫描范围2θ=5.0°~90.0°。采用高分辨率场发射扫描电子显微镜(SEM, GeminiSEM 300,德国蔡司公司,5 kV)、能谱仪(EDS)和透射电子显微镜(TEM,FEI Tecnai F30,荷兰飞利浦FEI公司,120.0 kV)观察样品的形貌、尺寸和元素特点。采用全自动比表面及孔隙度分析仪(2020 HD88,美国麦克仪器公司)进行氮气吸附-脱附性能测试,以分析样品的比表面积和中孔、大孔的孔径分布情况。采用有机元素分析仪(Vario EL Ⅲ,德国元素公司)以获得所制备样品的本体组成。采用X射线光电子能谱仪(XPS, Thermo ESCALAB 250XI,美国赛默飞世尔科技公司)分析样品的元素组成和化学态。采用紫外-可见漫反射光谱(UV-3600,日本岛津公司)分析样品对可见光的吸收性能。光致发光光谱(PL)光谱采用荧光分光光度计(F-7000,日本日立高新技术公司)测定,激发波长为350 nm。
通过在可见光照射下降解MB溶液来评价所制备样品的光催化活性。使用具有UV截止滤光器(λ > 420 nm)和500 W氙灯作为可见光光源的光催化反应仪(LAB500E4,北京中教金源)进行实验。在每个实验中,将50.0 mg光催化剂超声分散于100.0 mL浓度为30.0 mg·L-1的MB溶液中。在照射之前,将混合溶液避光搅拌3 h以建立吸附-解吸平衡。在光催化降解实验中,每隔30 min取样,通过0.22 μm的尼龙66滤膜过滤。使用紫外-可见分光光度计(DR2800,美国哈希公司)分析所得上清液,用MB在663 nm处的特征吸收峰来测定其吸光度。
催化剂的XRD图如图 1所示。球磨前后的SCN和BM-SCN-x具有相同的XRD衍射峰,表明球磨过程没有破坏SCN的晶体结构,但是可以观察到球磨后的BM-SCN-x在层状结构上发生了一定变化。SCN在2θ=13.1°处出现的衍射峰对应于g-C3N4的(100)晶面,与g-C3N4层内3-s-三嗪结构单元的周期性排列有关,而BM-SCN-x在此处的峰值不太明显,这可能归因于球磨后层的平面尺寸减小。SCN在2θ=27.4°附近的另一个强峰对应于共轭芳香族体系层间堆叠形成的(002)晶面,这个峰表明样品具有类石墨层状结构[17]。从图中可以看出,BM-SCN-x仍然保留了大部分的原始共轭结构,但是其(002)衍射峰的峰形稍微变宽,且衍射强度明显降低。引起峰形变宽的原因可能是样品的晶粒变小。使用Scherrer方程估算,得到SCN和BM-SCN-1的平均晶粒尺寸分别为35.2和28.6 nm。基于该计算,推断峰展宽的贡献主要是来自粒度的减小。另外,(002)衍射峰的峰值从27.4°向大角度产生了微小偏移(27.7°),说明球磨后得到的BM-SCN-x样品的层间堆叠变得更加紧密,层间距离减小[18]。由此可知,湿式球磨后的样品比干式球磨后的样品具有更薄的层状结构。
采用SEM观察催化剂的表面形貌和粒径大小,如图 2所示。热聚合法制备的硫掺杂g-C3N4表现出明显的体相形貌,由紧密片层堆积而成,样品粒径较大,为5.0~15.0 μm。干式球磨后的BM-SCN-0样品粒径减小但是不均匀,且可以清楚地看到破碎后的BM-SCN-0颗粒重新发生团聚,粒径为2.0~5.0 μm。与干式球磨相比,湿式球磨后得到的BM-SCN-1和BM-SCN-2样品粒径明显减小,分布得比较均匀,都在1.0 μm左右,并且发现有很多纳米级别的细小颗粒。在湿式球磨过程中,分散剂可以促使大颗粒裂纹扩展,同时阻止破碎后的小颗粒重新团聚,有利于球磨改性过程的进行[20]。由此可见,相比于干式球磨,湿式球磨可以有效地减小SCN的颗粒尺寸。

使用TEM进一步研究光催化剂样品的微观形貌,结果如图 3所示。由图 3a可见,未球磨的SCN呈现为较厚的块状物,表现出典型的二维层状结构,由大量表面致密的片层堆积而成。经过干式球磨后,BM-SCN-0的厚度和粒径明显变小(图 3b),但是从TEM图像中可以明显地看到许多凸出的较暗的部分,这归因于不同大小的硫掺杂g-C3N4颗粒的团聚现象。相比干式球磨,湿式球磨后得到的催化剂样品(图 3(c,d))颗粒尺寸更小,具有比较平坦的表面,且TEM图更加透明,说明在湿式球磨作用下获得了相对薄的g-C3N4层状结构,这与XRD得到的结果一致。

使用CHNS元素分析仪和EDS进行C、N、S三种元素的分析,以确定样品中硫的存在和分布情况,并分析球磨改性对催化剂本体组成的影响。选用图 2a里虚线框中的部分进行放大做EDS面扫,从图 4可以看出,硫元素在SCN表面上均匀分布。表 1总结了通过元素分析仪测得的所有样品的氮/碳原子比和硫的质量百分比,可以看到球磨前后硫掺杂g-C3N4的氮/碳原子比在1.53~1.50之间变化,而氮/碳原子比的理论值是1.33,这表明通过硫脲简单热缩聚制备的催化剂具有不完美的g-C3N4结构。因为在合成g-C3N4的聚合过程中会生成各种中间体,所以最终不可避免的形成一些典型的缺陷,而氨基物质是g-C3N4不完全聚合过程中产生的典型缺陷之一[19-21]。据报道,氨基物质可作为g-C3N4光催化剂中光生载流子的重组中心,使得光生电子-空穴更快地复合,导致光催化活性降低[22]。湿式球磨后BM-SCN-1的氮/碳原子比值从原来的1.52降至1.50,说明湿式球磨消除了g-C3N4表面上的部分氨基物质。这是由于球磨也是一种剥离的方法,这与Niu等[23]采用热氧化刻蚀法剥离g-C3N4而导致氮/碳原子比降低的报道一致。
 下载:
导出CSV
下载:
导出CSV
| Sample | Atomic ratio of N/C | Mass fraction of S / % |
| SCN | 1.52 | 0.35 |
| BM-SCN-0 | 1.53 | 0.39 |
| BM-SCN-1 | 1.50 | 0.33 |
| BM-SCN-2 | 1.51 | 0.19 |
图 5和表 2为SCN和BM-SCN-x样品的氮气吸附-脱附测试结果。由图 5a可知,湿式球磨后的样品在高的相对压力(P/P0 > 0.8)下具有高吸附容量,表明其存在丰富的介孔和大孔[24]。根据Brunauer-Emmett-Teller(BET)方法得到所有样品的比表面积,发现湿式球磨后的样品比表面积增大,其中BM-SCN-1的比表面积为35 m2·g-1,分别为SCN和BM-SCN-0的2.3和7.0倍。图 5b中湿式球磨后的样品在10.0~100.0 nm范围内的孔体积均较未球磨的SCN样品大,这说明湿式球磨使得硫掺杂g-C3N4产生了大量新的介孔。结果表明,干式球磨后的BM-SCN-0样品比表面积降低,孔隙率减少,这可能归因于干式球磨过程中破碎了的小颗粒重新团聚;相反,湿式球磨法能有效地提高硫掺杂g-C3N4的比表面积与孔隙率。对于光催化剂而言,介孔的数量越多,比表面积越高,可以提供更多的活性位点,增强对有机污染物的吸附和传输能力[25]。
 下载:
导出CSV
下载:
导出CSV
| Sample | BET surface area / (m2·g-1) | Average pore volume / (cm3·g-1) | Pore size / nm | Adsorption capacity for MB / (mg·g-1) |
| SCN | 15 | 0.01 | 11.32 | 9.50 |
| BM-SCN-0 | 5 | 0.06 | 14.20 | 8.69 |
| BM-SCN-1 | 35 | 0.13 | 13.84 | 32.24 |
| BM-SCN-2 | 26 | 0.08 | 11.62 | 29.88 |
| Operating parameters of absorption experiment: catalyst dosage 1.0 g·L-1, initial MB concentration of 40.0 mg·L-1 and reaction time of 48 h | ||||
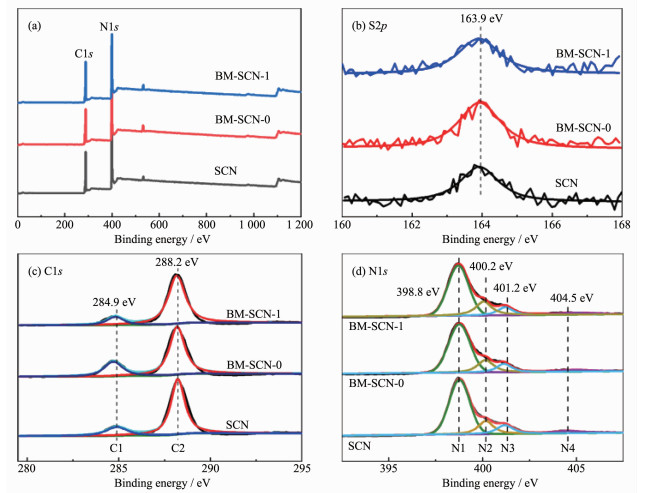
为了研究催化剂表面元素的化学态,对样品进行了XPS分析。图 6a是样品的XPS全谱图,结果表明所有样品都含有C、N、S元素,硫的质量百分比在球磨前后变化不大,都在0.22%左右。图 6b是S2p的高分辨图谱,163.9 eV处的峰归因于S通过取代g-C3N4晶格中的N原子形成了C-S键[11],球磨后的样品也基本上保留了硫掺杂的状态。图 6c是C1s高分辨图谱,在284.9(C1)和288.2 eV(C2)处具有2个明显的峰,分别对应于吸附在表面上的碳杂质(C-C和C=C)和g-C3N4三嗪环结构中的sp2杂化C原子(N-C=N)[12]。图 6d为N1s峰的拟合结果,最强峰位于398.8 eV(N1),来自三嗪结构单元(C-N=C)中与碳原子相连接的氮原子,而400.2 eV(N2)的弱峰是由sp3杂化的N-(C)3基团中的氮原子引起的。在401.2 eV(N3)处的另一个弱峰表明氨基官能团(C-N-H)的存在,而最弱的峰值为404.5 eV,归因于杂环中的电荷效应或正电荷定位[26]。图中没有发现对应C1s和N1s的结合能明显偏移,但从N1s峰和表 3里可以看到N3/N2物质的量比值的变化。通常,N-(C)3的存在是完全聚合的结果,而C-N-H的形成源自样品的不完全聚合,所以N2/N3物质的量比值的变化可代表硫掺杂g-C3N4的聚合度或氨基浓度的变化[19, 27]。与SCN相比,我们发现BM-SCN-1的N2/N3物质的量比值从1.42增加到1.97,BM-SCN-1的聚合度增加,这意味着湿式球磨消除了样品表面上未聚合的部分氨基,即缺陷浓度降低;但是BM-SCN-0的N2/N3物质的量比值减小到1.31,缺陷浓度反而稍微增加,这可能归因于干式球磨过程中固体表面之间的摩擦热更大导致温度过高而带来的副作用[16]。
 下载:
导出CSV
下载:
导出CSV
| Sample | Peak area of N2 | Peak area of N3 | Molar ratio of N2/N3 |
| SCN | 7 647.61 | 5 393.06 | 1.42 |
| BM-SCN-0 | 7 201.26 | 5 479.64 | 1.31 |
| BM-SCN-1 | 9 095.10 | 4 625.87 | 1.97 |
通过UV-Vis DRS研究催化剂的光吸收能力,图 7(a,b)显示的是球磨前后硫掺杂g-C3N4的紫外-可见漫反射吸收光谱图及其相应的禁带宽度图。如图所示,由于SCN粉末的颜色在球磨后从黄棕色变为淡黄色,球磨改性后的BM-SCN-x在可见光区域的光吸收能力减弱。SCN表现出在420~550 nm处的宽吸收峰,这归因于g-C3N4晶格中的硫掺杂[12]。样品SCN、BM-SCN-0和BM-SCN-1分别在450、420和430nm处显示出吸收带边缘,球磨后样品的吸收带边从450 nm最远可蓝移到420 nm,这种蓝移可能归因于量子尺寸效应[23],因为球磨可以有效地减小SCN的颗粒尺寸。对于半导体材料,其禁带宽度Eg可以由公式αhν=A(hν-Eg)n/2估算[28],其中α、h、ν、A、Eg分别为吸光系数、普朗克常数、光频率、能带常数和禁带宽度,n值由半导体催化剂的带间跃迁类型决定,直接跃迁n为1,间接跃迁n为4,SCN的n值为1[28]。由此方程计算出SCN和BM-SCN-x的带隙能量,结果依次为2.74、2.97、2.92、2.88eV。结合2张图可以看出,球磨改性后的样品禁带宽度均明显增大,导致其对可见光吸收的范围变窄,对可见光的利用效率降低[28];但是增加的禁带宽度有望在一定程度上提高其氧化还原能力,增强光催化活性[11]。

通过PL来表征光生载流子的迁移、传输和分离效率。由图 8可知,由于直接的带隙跃迁,所有样品的本征荧光衍射峰集中在440~455 nm左右,球磨后样品的PL峰位置发生蓝移,由SCN的455 nm迁移到BM-SCN-0的440 nm处,这归因于量子尺寸效应,并且与图 7b中禁带宽度的变化情况相吻合。另外,与SCN相比,干式球磨后BM-SCN-0样品的荧光强度高于SCN,光生电子与空穴的复合率增加;湿式球磨后BM-SCN-1和BM-SCN-2样品的荧光强度都低于SCN,光生电子与空穴的复合率降低。通常,作为表面缺陷的氨基基团可以捕获光激发的电子和空穴,导致在可见光照射期间电子-空穴的复合率更大[22]。PL强度的降低表明,由于表面缺陷的浓度降低,硫掺杂g-C3N4共轭体系的聚合度增加,湿式球磨后BM-SCN-1和BM-SCN-2样品中的电荷迁移率增强,从而抑制了电子-空穴的复合[29]。

球磨前后硫掺杂g-C3N4样品的光催化活性通过在可见光照射下降解有机染料MB来评价。如图 9a所示,湿式球磨得到的BM-SCN-1样品的效果最好,光照3 h后,湿式球磨改性后的BM-SCN-1样品对MB的降解率为91.5%,比未球磨的SCN样品提高32.8%,比干式球磨的BM-SCN-0样品提高50.9%。在光催化降解过程中,MB的光降解过程符合伪一级动力学特征[30]:
|
$ -\ln(C/C_0)=kt $ |
其中,C0代表吸附平衡后反应物的初始浓度;C代表催化反应t时间后反应物的浓度;k代表一级反应速率常数,可用来衡量光催化活性。如图 9b所示,SCN、BM-SCN-0、BM-SCN-1和BM-SCN-2的k值分别为0.308、0.167、0.764、0.574 h-1。可见,BM-SCN-1的降解速率最快,分别比未球磨和干式球磨后的样品提高了1.5和3.6倍。这可能是由于BM-SCN-1比表面积相对较大,能够暴露更多的反应活性位点,染料分子较容易吸附到催化剂表面上,从而与活性物种更充分地反应[31]。另外,湿式球磨后的BM-SCN-1表面缺陷减少,聚合度增加,这有利于提高其光催化活性[22]。

在光催化降解有机污染物的反应中,光催化剂的循环利用稳定性也十分重要。图 10a为5次循环实验后样品的XRD图,对比图 1可知,光催化反应对球磨前后的硫掺杂g-C3N4的晶体结构影响不大,说明制备的光催化剂具有良好的结构稳定性。将SCN和BM-SCN-x催化剂在同样的实验条件下循环使用5次,如图 10b所示,SCN和BM-SCN-x样品的催化活性都轻微降低,这可能是由于有机物分子吸附在材料表面,消耗了部分吸附位点和活性中心[32]。另外,SCN和BM-SCN-x样品在5次循环后的光催化活性都在初始活性的87.2%~90.4%范围内,这表明硫掺杂g-C3N4在球磨前后的光催化稳定性没有发生明显变化。

以硫脲为前驱体,通过热聚合法制备了原位硫掺杂g-C3N4,采用球磨法对硫掺杂的g-C3N4作进一步的改性处理。与未球磨和干式球磨后的样品相比,湿式球磨后的BM-SCN-1粒径大大减小且具有较宽的介孔分布,比表面积(35 m2·g-1)较未球磨的SCN提高了1.3倍,反应活性位点数量增加。由于量子尺寸效应,湿式球磨后的硫掺杂g-C3N4能带隙增大,更宽的禁带赋予载流子更强的氧化还原能力。另外,经湿式球磨改性后,其表面缺陷的浓度降低,这促进了光生载流子的有效分离和转移,有利于电子-空穴复合率的降低。从可见光催化降解亚甲基蓝的实验结果可知,湿式球磨后的硫掺杂g-C3N4光催化性能明显提高,其中BM-SCN-1性能最优,降解速率分别比未球磨的样品和干式球磨后的样品提高了1.5和3.6倍。本研究所采用的湿式球磨法将有望为其他高效光催化剂的绿色改性研究提供新的策略。
Mamba G, Mishra A K. Appl. Catal. B, 2016, 198:347-377 doi: 10.1016/j.apcatb.2016.05.052
Cao S W, Low J X, Yu J G, et al. Adv. Mater., 2015, 27(13):2150-2176 doi: 10.1002/adma.201500033
Ye S, Wang R, Wu M Z, et al. Appl. Surf. Sci., 2015, 358:15-27 doi: 10.1016/j.apsusc.2015.08.173
Chen Y, Jia G, Hu Y F, et al. Sustainable Energy Fuels, 2017, 1(9):1875-1898 doi: 10.1039/C7SE00344G
Xu B T, Ahmed M B, Zhou J L, et al. Sci. Total Environ., 2018, 633:546-559 doi: 10.1016/j.scitotenv.2018.03.206
Zhu J J, Xiao P, Li H L, et al. ACS Appl. Mater. Interfaces, 2014, 6(19):16449-16465 doi: 10.1021/am502925j
Patnaik S, Martha S, Parida K M. RSC Adv., 2016, 6(52):46929-46951 doi: 10.1039/C5RA26702A
Jiang L B, Yuan X Z, Pan Y, et al. Appl. Catal. B, 2017, 217:388-406 doi: 10.1016/j.apcatb.2017.06.003
Zhang S, Gu P C, Ma R, et al. Catal. Today, 2019, 335:65-77 doi: 10.1016/j.cattod.2018.09.013
Zhou L, Zhang H Y, Sun H Q, et al. Catal. Sci. Technol., 2016, 6(19):7002-7023 doi: 10.1039/C6CY01195K
Liu G, Niu P, Sun C H, et al. J. Am. Chem. Soc., 2010, 132(33):11642-11648 doi: 10.1021/ja103798k
Wang K, Li Q, Liu B S, et al. Appl. Catal. B, 2015, 176:44-52
Cao L, Wang R, Wang D X. Mater. Lett., 2015, 149:50-53 doi: 10.1016/j.matlet.2015.02.119
Hong J D, Xia X Y, Wang Y S, et al. J. Mater. Chem., 2012, 22(30):15006-15012 doi: 10.1039/c2jm32053c
Lu C H, Zhang P, Jiang S J, et al. Appl. Catal. B, 2017, 200:378-385 doi: 10.1016/j.apcatb.2016.07.036
James S L, Adams C J, Bolm C, et al. Chem. Soc. Rev., 2012, 41(1):413-447 doi: 10.1039/C1CS15171A
Cai Q F, Shen J C, Feng Y, et al. J. Alloys Compd., 2015, 628:372-378 doi: 10.1016/j.jallcom.2014.12.013
Zhu K X, Wang W J, Meng A L, et al. RSC Adv., 2015, 5(69):56239-56243 doi: 10.1039/C5RA09040G
Kim J S, Oh J W, Woo S I. Catal. Today, 2017, 293:8-14
McMahon B W, Perez J P L, Yu J, et al. ACS Appl. Mater. Interfaces, 2014, 6(22):19579-19591 doi: 10.1021/am503845s
Wu P, Wang J R, Zhao J, et al. J. Mater. Chem. A, 2014, 2(47):20338-20344 doi: 10.1039/C4TA04100C
Lin L H, Ou H H, Zhang Y F, et al. ACS Catal., 2016, 6(6):3921-3931 doi: 10.1021/acscatal.6b00922
Niu P, Zhang L L, Liu G, et al. Adv. Funct. Mater., 2012, 22(22):4763-4770 doi: 10.1002/adfm.201200922
Li Y F, Jin R X, Xing Y, et al. Adv. Energy Mater., 2016, 6(24):1601273-1601277 doi: 10.1002/aenm.201601273
侯建华, 蔡瑞, 沈明, 等.无机化学学报, 2018, 34(3):467-474HOU Jian-Hua, CAI Rui, SHEN Ming, et al. Chinese J. Inorg. Chem., 2018, 34(3):467-474
Lin Z Z, Wang X C. Angew. Chem. Int. Ed., 2013, 52(6):1735-1738 doi: 10.1002/anie.201209017
Li X B, Hartley G, Ward A J, et al. J. Phys. Chem. C, 2015, 119(27):14938-14946 doi: 10.1021/acs.jpcc.5b03538
李娜, 王茗, 赵北平, 等.无机化学学报, 2016, 32(6):1033-1040 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20160614&journal_id=wjhxxbcnLI Na, WANGMing, ZHAO Bei-Ping, et al. Chinese J. Inorg. Chem., 2016, 32(6):1033-1040 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20160614&journal_id=wjhxxbcn
Guo Y F, Li J, Yuan Y P, et al. Angew. Chem. Int. Ed., 2016, 55(47):14693-14697 doi: 10.1002/anie.201608453
Chen W, Liu T Y, Huang T, et al. Nanoscale, 2016, 8(6):3711-3719 doi: 10.1039/C5NR07695A
王晓雪, 高建平, 赵瑞茹, 等.无机化学学报, 2018, 34(6):1059-1064 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20180607&journal_id=wjhxxbcnWANG Xiao-Xue, GAO Jian-Ping, ZHAO Rui-Ru, et al. Chinese J. Inorg. Chem., 2018, 34(6):1059-1064 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20180607&journal_id=wjhxxbcn
祝凯, 欧阳杰, 刘家满, 等.无机化学学报, 2019, 35(6):1005-1012 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20190609&journal_id=wjhxxbcnZHU Kai, OUYANG Jie, LIU Jia-Man, et al. Chinese J. Inorg. Chem., 2019, 35(6):1005-1012 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20190609&journal_id=wjhxxbcn

图 2 (a) SCN、(b) BM-SCN-0、(c) BM-SCN-1和(d) BM-SCN-2样品的SEM图
Figure 2 SEM images of (a) SCN, (b) BM-SCN-0, (c) BM-SCN-1 and (d) BM-SCN-2

图 3 (a) SCN、(b) BM-SCN-0、(c) BM-SCN-1和(d) BM-SCN-2样品的TEM图
Figure 3 TEM images of (a) SCN, (b) BM-SCN-0, (c) BM-SCN-1 and (d) BM-SCN-2

图 5 SCN和BM-SCN-x的(a)氮气吸附-脱附等温线和(b)孔径分布曲线
Figure 5 (a) Nitrogen adsorption-desorption isotherms and (b) pore size distribution curves of SCN and BM-SCN-x

图 6 SCN和BM-SCN-x的(a) XPS全谱图和(b) S2p, (c) C1s和(d) N1s的高分辨光谱图
Figure 6 (a) XPS survey spectra (a) and high-resolution XPS spectra of (b) S2p, (c) C1s and (d) N1s for SCN and BM-SCN-x

图 7 SCN和BM-SCN-x样品的(a) UV-Vis DRS及(b) (αhν)2-hν曲线
Figure 7 (a) UV-Vis DRS and (b) (αhν)2-hν curves of SCN and BM-SCN-x

图 8 SCN和BM-SCN-x光催化剂的光致发光光谱
Figure 8 Photoluminescence spectra of SCN and BM-SCN-x samples

图 9 SCN和BM-SCN-x催化剂的(a)光降解反应和(b)降解速率
Figure 9 (a) Photocatalytic reaction under visible light and (b) degradation rates by SCN and BM-SCN-x catalysts

图 10 五次循环实验后SCN和BM-SCN-x样品的(a) XRD图和(b)循环降解曲线
Figure 10 (a) XRD patterns after 5 cycles and (b) cycling degradation activity of SCN and BM-SCN-x samples
表 1 SCN和BM-SCN-x样品的氮/碳原子比和硫含量
Table 1. Nitrogen/carbon atomic ratio and amount of sulfur in the SCN and BM-SCN-x samples
| Sample | Atomic ratio of N/C | Mass fraction of S / % |
| SCN | 1.52 | 0.35 |
| BM-SCN-0 | 1.53 | 0.39 |
| BM-SCN-1 | 1.50 | 0.33 |
| BM-SCN-2 | 1.51 | 0.19 |
 下载: 导出CSV
下载: 导出CSV
表 2 SCN和BM-SCN-x样品的比表面积、平均孔体积、孔径和对MB的吸附容量
Table 2. Surface area, average pore volume, pore size, adsorption capacity for MB of the SCN and BM-SCN-x samples
| Sample | BET surface area / (m2·g-1) | Average pore volume / (cm3·g-1) | Pore size / nm | Adsorption capacity for MB / (mg·g-1) |
| SCN | 15 | 0.01 | 11.32 | 9.50 |
| BM-SCN-0 | 5 | 0.06 | 14.20 | 8.69 |
| BM-SCN-1 | 35 | 0.13 | 13.84 | 32.24 |
| BM-SCN-2 | 26 | 0.08 | 11.62 | 29.88 |
| Operating parameters of absorption experiment: catalyst dosage 1.0 g·L-1, initial MB concentration of 40.0 mg·L-1 and reaction time of 48 h | ||||
下载: 导出CSV
表 3 SCN和BM-SCN-x样品的N2/N3物质的量比值
Table 3. Molar ratio of N2/N3 for SCN and BM-SCN-x samples
| Sample | Peak area of N2 | Peak area of N3 | Molar ratio of N2/N3 |
| SCN | 7 647.61 | 5 393.06 | 1.42 |
| BM-SCN-0 | 7 201.26 | 5 479.64 | 1.31 |
| BM-SCN-1 | 9 095.10 | 4 625.87 | 1.97 |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: