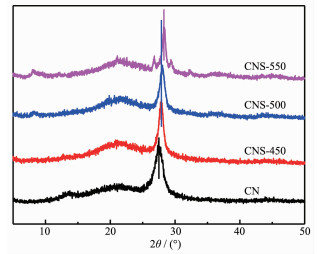
图 1.
CN及CNS样品的XRD图
Figure 1.
XRD patterns of CN and CNS samples

光催化技术可以在温和的条件下实现太阳能转化,因此利用光催化技术分解水制取氢气是发展清洁能源的一条有效途径[1]。非金属聚合物半导体石墨相氮化碳(g-C3N4)因其制备原料丰富、稳定无污染等优点在光催化领域极具广阔的发展应用空间[2-4]。然而,无定型聚合物结构固有的较低电子传输特性限制了其在光催化性领域的实际应用。
截至目前,大量的研究工作针对g-C3N4的结构优化改性以提高其光催化性能,包括金属/非金属掺杂[5-6]、半导体复合[7]、形貌控制[8]等,其中,简单后热处理可进一步丰富g-C3N4的多孔性,显著增大其比表面积,同时引入表面氮缺陷位,可在一定程度上提高其光催化性能[9]。尽管如此,对g-C3N4进行后热处理,进一步降低了材料的聚合度,增加了结构无序性,限制了光生载流子有效传输提高的幅度[10-11]。
早在2008年,Thoms等[12]以LiCl/KCl混合熔盐为介质,离子热聚合二氰二胺制备得到结晶相g-C3N4。近年来,Dontsova等[13]以含ZnCl2混合熔盐为介质,热聚合三聚氰胺得到高比表面积的g-C3N4,具有优异的CO2/N2吸附特性。所以熔盐离子热聚合可制备得到高度聚合的g-C3N4材料[14]。然而直接聚合富氮单体分子所得产物为聚三嗪结构(PTI),光催化活性不理想[15]。比起PTI结构,七嗪结构的g-C3N4具有更宽的可见光吸收和更高的热稳定性,有利于光催化反应的进行[16]。因此,结合以上2种途径,以g-C3N4为原料进行熔盐后热处理有望降低结构缺陷,有利于光催化性能的提高。
本研究以二氰二胺为原料热聚合法制备g-C3N4材料,并在空气中对其进行不同温度下的二次后热处理。以LiCl/KCl混合熔盐为介质二次热处理得到高聚合度的g-C3N4材料。同时对比常规直接后热处理过程,对产物的结构进行了详细表征和分析。通过光催化分解水制氢来评价催化剂的活性。研究结果表明,熔盐离子热后热处理得到的光催化材料表现出更高的可见光催化制氢活性和稳定性。
分别取4 g尿素、4 g二氰二胺溶于20 mL超纯水中,80 ℃油浴搅拌使原料充分混合,充分干燥后在马弗炉中550 ℃高温煅烧4 h。所得产物记为CN0。将所得固体粉末在马弗炉中450、500、550 ℃下直接二次煅烧4 h,样品记为CN-450、CN-500、CN-550。以下没有特殊说明,CN指CN-500。
将1.2 g CN0粉末与6.6 g KCl及5.4 g LiCl在研钵中充分研磨混合。分别在马弗炉中450、500、550 ℃下煅烧4 h;冷却至室温,超纯水洗涤3~5次,透析袋透析96 h,去除无机盐离子,过滤收集,最后在60 ℃下真空干燥。样品分别标记为CNS-450、CNS-500和CNS-550。
采用XRD (D/max 2500PC,日本理学株式会社)分析材料成分,测试电压及电流分别为40 kV和30 mA,Cu Kα射线(波长为0.150 4 nm)辐射,扫描范围为5°~50°;通过TEM(JEM-2100,日本理学株式会社)观察样品形貌和晶相,工作电压为200 kV;采用BET(Brunauer-Emmett-Teller)比表面积分析仪(JW-BK122W,精微高博)进行N2吸附-脱附测试,分析样品的比表面积及孔径分布;采用UV-Vis(UV-2550,日本Shimadzu公司)表征样品的光吸收性质,BaSO4为标准物质,扫描范围200~800 nm;通过ESCALAB 250(美国Thermo Fisher Scientific公司)测定材料X射线光电子能谱(XPS);采用QuantaMasterTM40(美国PTI公司)考察催化剂的荧光发光性能;利用电化学工作站(CHI660E,上海辰华仪器有限公司)表征催化剂的光电化学性质。
50 mg催化剂分散于100 mL含有10%(V/V)三乙醇胺的水溶液中,将100 μL氯铂酸水溶液(H2PtCl6·6H2O,0.04 g·mL-1)加入到上述溶液中并搅拌30 min。系统抽真空,使用300 W氙灯光源(400 nm cut滤光片)进行光照,3%(w/w)的Pt原位光沉积在催化剂表面。每隔20 min通过气相色谱(TCD检测器,N2为载气)在线自动取样分析H2的产生量。在连续长时间光照测试过程中,催化剂用量25 mg。
图 1为直接煅烧和熔盐离子热后热处理制备的CN和CNS的XRD图。由图中可以看出,CN具有典型的石墨相氮化碳XRD衍射峰,分别位于27.4°和13.3°,对应石墨层间堆积的长程有序(002)和面内重复单元堆积(100)衍射峰[17]。经熔盐离子热在不同温度下处理后得CNS的XRD衍射峰发生明显变化。CNS在13.3°处衍射峰强度降低至几乎消失。当热处理温度提高至500 ℃,在8.1°处出现新的衍射峰,表明面内聚合杂环间距增大,这可能与面内共轭骨架的充分缩合有关。同时,CN在27.4°处的((002)面,d=0.324 nm)衍射峰逐步向高角度偏移,对应CNS-450 27.8°(d=0.320 nm)、CNS-500 27.9°(d=0.318 nm)、CNS-550 28.3°(d=0.315 nm),层间距的缩小说明二维层间堆积致密性增加,源于高温液相介质有利于层状CN堆积重组。另外,从(002)面衍射峰逐渐缩小的半峰宽可以看出,经过熔盐热处理后,CNS的石墨相聚合度逐步提高。长程有序度的提高对促进聚合物结构中载流子的传输具有重要意义。然而CNS-550在主峰附近新产生其他弱衍射峰,可能来源于高温下结构变形形成新的衍生聚合晶相。
图 2为所得催化剂样品的SEM和TEM图。相对于CN样品,CNS具有更小的颗粒尺寸,这可能是由于熔盐离子间作用的结果,与前期报道相一致[18]。从TEM图中可以看出,CNS样品为薄片状结构。高分辨透射电镜(HRTEM)图中可以看到清晰的晶格条纹,对应晶面间距0.32 nm,与XRD测试结果一致,证明了CNS具有高的结晶聚合度[19]。因此,经过熔盐离子热处理后的CNS样品颗粒尺寸降低,预示具有更大的比表面积,增加的表面活性位有利于多相催化反应的进行。

图 3a为CN及CNS样品的N2吸附-脱附曲线,为典型的Ⅳ型等温线。随着离子热处理温度的提高,在P/P0=0.6~0.9区间出现明显的H3型滞回线,对应介孔分布。经过二次热处理,CN的比表面积为18 m2·g-1。相对CN,CNS具有显著增大的比表面积,其中CNS-500具有最大值,提高至110 m2·g-1。图 3b为催化剂相应的孔分布曲线。从图中可以看出,相对于CN,CNS样品在5~30 nm范围内的孔体积显著增大,其中CNS-500增大最为显著。多孔性的增加对促进光催化反应具有重要作用[20]。

图 4为CN和CNS样品的FT-IR图谱。相比于CN,CNS样品在3 000~3 400 cm-1范围内的峰宽降低,主要集中在3 400 cm-1附近,对应于表面吸附水引起的O-H吸收峰,而在3 000~3 300 cm-1范围内的氨基吸收峰强度降低,说明离子热处理有利于消除表面氨基[21]。另外,CNS在2 200 cm-1附近出现新的吸收峰,对应于未完全聚合末端氰基,说明熔盐离子的参与使得部分面内聚合单元被“剪断”,这与XRD表征中(100)面特征峰向低角度偏移是一致的[22]。除此之外,在500~4 000 cm-1范围,CNS样品显示出与CN类似的红外光谱,说明离子热处理不改变催化剂的基本共轭键合结构。
采用XPS技术表征催化剂表面元素状态。图 5a为CNS-500的元素总图谱,该图显示除了C、N、O外,还含有少量K元素,结合图 5b中归属于K+的292.8和295.5 eV的双峰,说明少量K+在熔盐离子热过程中被引入框架结构[23]。由于K+的电荷补偿效应,引起催化剂末端聚合不完全,表面产生氰基,这与FT-IR表征结果相一致。

图 5(c, d)分别对CN和CNS-500的C1s和N1s的高分辨拟合图谱。C1s谱主要拟合为3个峰,分别位于284.6、286.5和288.0 eV。其中,284.6 eV归属于石墨碳(C1),而286.5和288.0 eV则分别属于少量C-O(C2)的存在和C-N杂环中的sp2杂化碳(N-C=N)(C3),而C3构成聚合物氮化碳的主要骨架。对比发现,CNS-500中C3在C元素中的含量为82%,低于CN中C3在C元素中的含量98%。说明熔盐热处理后引起sp2 C含量降低。N1s谱同样可拟合为3个峰,分别位于398.5、399.5和400.9 eV。其中,最强峰398.5 eV属于聚三嗪结构中的sp2杂化N(N1)。399.5 eV峰属于桥连N(N-(C)3)(N2)。400.9 eV峰说明催化剂表面存在末端氨基基团(C-N-H) (N3)[24]。对比发现,N1s在N元素中的含量由CN样品中的72%降低至CNS-500样品中的68%,证明了sp2 N的降低。同时,CNS-500样品中N2和N3峰位均向低位移动,说明具有增强的电荷密度。因此,离子热处理后的样品,增加了CN共轭sp2 N缺陷,降低了表面氨基,产生更强吸电子基团-C≡N,增强表面电荷,有利于光催化还原反应的进行。图 5(e)为CNS-500的结构示意图。
图 6为经过不同温度后热处理后所得CN和CNS样品的紫外-可见吸收光谱。随着热处理温度的提高,CN的光吸收带边稍微蓝移,这归因于热处理后引起的粒子尺寸减小[25]。相反的,CNS的光吸收带边却发生显著红移,说明除了量子尺寸效应,离子热处理后催化剂本身的高聚合度对光吸收改善起到主要作用。同时,对比相同后热处理温度的样品,CNS样品的光吸收带边发生显著红移,CN和CNS-500的吸收带边分别位于445和470 nm,根据公式Eg=1 240/λ(λ为吸收带边波长),对应带隙宽度(Eg)分别为2.78和2.64 eV。同时,CNS样品在紫外-可见光区的光吸收强度显著增强。光吸收性能的增强和吸收带边的红移意味着催化剂更容易被可见光激发,更易产生光生载流子。
图 7为CN和CNS的稳态荧光发射谱。在400 nm激发光下,CN在450 nm附近具有明显的宽光致发光光谱(PL)发射峰,来源于激发电子-空穴复合[26]。在熔盐热处理后,CNS-500的PL峰强度显著降低,在470 nm附近显示弱PL发射峰,说明CNS样品中激发态电子-空穴对的复合率大幅度降低[27]。大量“自由态”光生电子的存在有助于光催化还原分解水制氢的催化活性提高。

进一步采用瞬态PL方法拟合计算催化剂的PL寿命(图 7b)。经二阶导数拟合计算[28],CN与CNS-500的平均PL寿命分别为2.28和1.92 ns。可以看出,经过熔盐二次热处理后,催化剂的PL寿命显著延长,其中熔盐离子热处理后得到的产物PL寿命进一步缩短,证明了加速的光生载流子流动性[29]。
图 8为CN和CNS-500的电化学表征测试结果。图中可以看出,相比于CN-500,经过熔盐离子热处理的CNS-500具有更高的光电流响应。而且,经过多次“开-关”光照反应,CNS-500具有更加稳定的光电流产生,无衰减趋势。同时,交流阻抗谱也同样显示,CNS-500具有更小的阻抗曲线半径,表示更低的阻抗值。电化学表征结果表明,熔盐离子热后处理的样品具有更加优化的光电响应[30]。
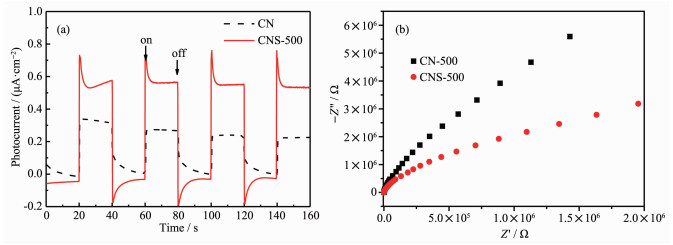
图 9a为所得样品的产氢活性图。所有催化剂均随着光照时间的延长产氢量持续增加,说明了催化剂对光具有延续响应性。由图中可以看出,经过后热处理后的CNS催化剂的产氢活性与同温度直接热处理样品相比,均有显著提高,其中CNS-500的制氢活性最高,产氢速率达192 μmol·h-1 (3 840 μmol·h-1·g-1),是CN的3.84倍。

为进一步验证催化剂的活性稳定性,对CNS-500催化剂进行连续产氢测试(图 9b)。CNS-500催化剂在连续的20 h内能够连续稳定的产生氢气,且基本保持线性增长趋势。说了熔盐离子热后热处理得到的催化剂具有较好的光催化产氢稳定性。
同时,对反应后的CNS-500样品进行了FT-IR和HRTEM测试,结果如图 10所示。经过2 h的催化反应后,其基本C-N共轭骨架基本不发生变化,依然保持缺陷位的存在(-C≡N基团)。从HRTEM图片中依然清晰可见(002)面层间堆积晶格条纹,说明其高聚合度稳定保持。因此,经过催化制氢反应后,CNS-500保持较高的结构稳定性。

后热处理能够调控氮化碳催化剂的聚合结构,同时引入结构缺陷,改变产物的聚合度。与传统直接热处理方法相比,熔盐离子热后热处理可显著提高氮化碳的聚合结晶度,缩小层间距,长程有序度得到优化改善。同时,熔盐离子热可大幅扩展催化剂的可见光吸收,增大产物孔隙率和比表面积。适宜的热处理温度有利于改善催化剂的微观聚合结构,共轭杂环中引入强吸电子基团,有利于电荷重新分布。经过500 ℃熔盐后处理,所得催化剂在可见光下的制氢活性大幅度增强。
Wang F F, Li Q, Xu D S. Adv. Energy Mater., 2017, 7(23):1700529 doi: 10.1002/aenm.201700529
Zhao A W, Sun Y J, Dong F. Nanoscale, 2015, 7(1):15-37 http://www.ncbi.nlm.nih.gov/pubmed/25407808
Wen J Q, Xie J, Chen X B, et al. Appl. Surf. Sci., 2017, 391:72-123 doi: 10.1016/j.apsusc.2016.07.030
崔言娟, 王愉雄, 王浩, 等.化学进展, 2016, 28(4):428-437 http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201604003.htmCUI Yan-Juan, WANG Yu-Xiong, WANG Hao, et al. Progress in Chemistry, 2016, 28(4):428-437 http://www.cnki.com.cn/Article/CJFDTotal-HXJZ201604003.htm
Jiang L B, Yuan X Z, Liang J, et al. Appl. Catal. B, 2017, 217:388-406 doi: 10.1016/j.apcatb.2017.06.003
Su Q, Yao X Q, Cheng W G, et al. Green Chem., 2017, 9(13):2957-2965
Song Y H, Gu J M, Xia K X, et al. Appl. Surf. Sci., 2019, 467:56-64
Wu M, Zhang J, He B B, et al. Appl. Catal. B, 2019, 241:159-166 doi: 10.1016/j.apcatb.2018.09.037
Cui Y J, Wang H, Yang C F, et al. Appl. Surf. Sci., 2018, 441:621-630 doi: 10.1016/j.apsusc.2018.02.073
Yang C F, Teng W, Song Y H, et al. Chin. J. Catal., 2018, 39(10):1615-1624 doi: 10.1016/S1872-2067(18)63131-6
Teng Z Y, Lv H Y, Wang C Y, et al. Carbon, 2017, 113:63-75 doi: 10.1016/j.carbon.2016.11.030
Bojdys M J, Müller J O, Antonietti M, et al. Chem. Eur. J., 2008, 14(24):8177-8182 doi: 10.1002/chem.200800190
Fettkenhauer C, Weber J, Antoniettia M, et al. RSC Adv., 2014, 4(77):40803-40811 doi: 10.1039/C4RA08236B
Chen Z P, Savateev A, Pronkin S, et al. Adv. Mater., 2017, 29(32):1700555 doi: 10.1002/adma.201700555
Tian L, Li J Y, Liang F, et al. Appl. Catal. B, 2018, 225:307-313 doi: 10.1016/j.apcatb.2017.11.082
Lin L H, Ou H H, Zhang Y F, et al. ACS Catal., 2016, 6(6):3921-3931 doi: 10.1021/acscatal.6b00922
Wang C, Fan H Q, Ren X H, et al. ChemSusChem, 2018, 11(4):700-708 doi: 10.1002/cssc.201702278
Ou H H, Lin L H, Zheng Y, et al. Adv. Mater., 2017, 29(22):1700008(13 Pages)
Zhang G G, Li G S, Lan Z A, et al. Angew. Chem. Int. Ed., 2017, 56(43):13630-13634
Zhou Y, Lv W H, Zhu B L, et al. ACS Sustainable Chem. Eng., 2019, 7(6):5801-5807 doi: 10.1021/acssuschemeng.8b05374
Wirnhier E, Dblinger M, Gunzelmann D. Chem. Eur. J., 2011, 17(11):3213-3221 doi: 10.1002/chem.201002462
Yu H J, Shi R, Zhao Y X, et al. Adv. Mater., 2017, 29(16):1605148(54 Pages)
Mo Z, Xu H, Chen Z G, et al. Appl. Catal. B, 2019, 241:452-460 doi: 10.1016/j.apcatb.2018.08.073
Oh J H, Yoo R J, Kim S Y, et al. Chem. Eur. J., 2015, 21(16):6241-6246 doi: 10.1002/chem.201406151
Zheng D D, Huang C J, Wang X C. Nanoscale, 2015, 7(2):465-470 doi: 10.1039/C4NR06011C
Shi Y H, Huang J H, Zeng G M, et al. J. Colloid Interface Sci., 2018, 531:433-443 doi: 10.1016/j.jcis.2018.07.079
Song Y H, Xia K X, Gong Y M, et al. Dalton Trans., 2018, 47(41):14706-14712 doi: 10.1039/C8DT03161D
Ran J R, Ma T Y, Gao G P, et al. Energy Environ. Sci., 2015, 8(12):3708-3717 doi: 10.1039/C5EE02650D
Liu J Y, Fang W J, Wei Z D, et al. Appl. Catal. B, 2018, 238:465-470 doi: 10.1016/j.apcatb.2018.07.021
Meng S G, Ye X J, Ning X F, et al. Appl. Catal. B, 2016, 182:356-368 doi: 10.1016/j.apcatb.2015.09.030

图 2 (a, b) CN和CNS-500的SEM图; (c, d) CNS-500的TEM及HRTEM图
Figure 2 (a, b) SEM images of CN and CNS-500; (c, d) TEM and HRTEM images of CNS-500

图 3 CN及CNS样品的N2吸附-脱附图(a)及相应的孔分布曲线(b)
Figure 3 N2 adsorption-desorption isotherms (a) and the corresponding pore-size distribution (b) of CNS samples

图 5 CNS-500的XPS总谱(a)和K2p谱(b); (c, d) CN和CNS-500的C1s和N1s谱; (e) CNS-500的结构示意图
Figure 5 Survey (a) and K2p (b) XPS spectra of CNS-500; (c, d) C1s and N1s spectra of CN and CNS-500; (e) Structural diagram of CNS-500

图 7 CN和CNS的稳态PL谱(a)和瞬态PL谱(b)
Figure 7 Steady-state PL (a) and transient PL spectra of CN and CNS samples

图 8 CN-500和CNS-500样品的(a)瞬时光电流和(b)阻抗图谱
Figure 8 Transient photocurrent response (a) and impedance spectroscopy (b) of CN and CNS-500

图 9 (a) CN和CNS样品的产氢活性; (b) CNS-500的连续产氢活性
Figure 9 (a) H2 evolution of CN and CNS samples under visible light irradiation; (b) Stability test of H2 evolution for CNS-500

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:



 下载:
下载: