
图 1.
经典结构的MOFs种类举例
Figure 1.
Examples of MOFs with classical structures

经济的快速增长促使能源的使用需求不断攀升,进而加剧了化石燃料燃烧带来的各种环境及资源短缺问题,寻求可再生的新能源已成为人们面临的迫切任务[1-3]。太阳能、风能等资源的地域性和间歇性引发了人们对环境友好的能源存储和转换器件的广泛关注,比如锂离子电池、超级电容器、电分解水制氢等技术[4-7]。为此,众多国内外研究人员致力于探索和开发具有高容量和高效率的能源存储和转换装置,而其中最为关键的是寻求清洁、环保的高性能材料[8-9]。
多孔碳材料是一种资源丰富且成本低廉的环境友好型材料,一般具有大的比表面积、高的导电性、优异的热和化学稳定性等优点,这促使多孔碳材料在储能器件和电催化氧还原反应中均显示出潜在的应用前景[10-11]。目前,合成多孔碳材料的常用方法是高温热解有机分子或生物质材料[12-13]。为得到高孔隙率的碳材料,这类方法往往要经过后续的物理/化学活化处理,或在合成中使用大量的模板,使得制备过程变得复杂繁琐,而且难于控制碳材料的孔径和颗粒尺寸分布,为实现其最佳性能及探索其作用机制带来困扰[14-15]。
近来,金属-有机框架(metal-organic frame-works,MOFs)衍生的碳材料引起了科研人员的广泛重视。MOFs是一种由含氮或氧等基团的有机配体与金属离子或金属离子簇通过配位键连接而成的具有规则孔道结构的三维网络框架材料[16]。作为一种新型的多孔有机-无机杂化材料,MOFs可以通过选择适宜的有机配体和金属来调控其组成和结构,因此MOFs材料具有组成及结构丰富、孔道多样和孔隙率高等特点[17]。目前,已知的MOFs大约有20 000多种,主要通过直接溶液合成、水热、溶剂热、微波或超声辅助及电化学合成等多种方法制备,其中ZIF-8、ZIF-67、MOF-5、HKUST-1、MIL-101、UiO-66等一些经典结构的MOFs(图 1),因合成方法简单、颗粒尺寸易调、孔隙率高等优点经常被作为优良的前驱体或模板。以MOFs为前驱体,在惰性气氛条件下制备的碳材料有效保留了MOFs前驱体的孔结构以及大比表面积等特性,且具有导电性强及稳定性高等突出优点[18-20]。此外,通过选用合适的MOFs前驱体,所制备的碳材料可以实现原位多种杂原子掺杂(如N、P、S、B等)。杂原子在空间上的规则排列和均匀分布,及可调控的微观形貌和颗粒尺寸使MOFs成为制备多孔碳材料的优良模板和前驱体[21-24]。因此,MOFs衍生的多孔碳材料发展迅速,并被尝试应用于储能器件及氧还原反应等诸多领域[25-29]。
有趣的是,最近研究发现,相比于由MOFs制备的实心型多孔碳材料,MOFs衍生的多孔壳层结构碳材料,即中空多孔碳材料,在二次电池、超级电容器、电催化等领域显示出更为优异的电化学性能[30-31]。主要是因为中空结构的碳材料比实心型的碳材料具有更低的密度、更多的活性位点暴露,可与反应介质充分接触。此外,中空材料的内部空腔又可提高活性物质负载量,使物质的扩散更为便利,在缩短传输路径的同时为活性物质的体积膨胀提供缓冲空间,进而有效地缓解活性材料在电化学反应过程中的形变,提高材料的整体稳定性[32]。MOFs衍生的中空多孔碳材料的独特结构优势引起了越来越多科研人员的研究兴趣,故而这类碳材料被尝试应用于多个研究领域[33-36]。
以MOFs为前驱体制备的中空多孔碳材料纳米颗粒可呈现十二面体、立方体、球形及管状结构等多种形貌,其颗粒尺寸及空腔大小集中在几十纳米到几百纳米不等,制备方法多采用模板辅助、界面组装、化学蚀刻及高温热处理等[37-40]。由此制备的中空碳材料具有丰富的结构多样性,其形貌、孔道、空腔大小等均可调控,极大地扩宽了它们的应用范围,尤其在电化学相关的领域显示出巨大的优势。因此,本文将主要概述MOFs衍生的中空多孔碳材料在储能器件及氧还原反应中应用的最新研究进展,包括以MOFs为前驱体制备中空多孔碳材料的方法及中空多孔碳材料的结构组成,并对其在锂离子电池、锂硫/硒电池、钠离子电池、超级电容器、电催化氧还原等领域的应用进行了论述。最后,对MOFs衍生的中空多孔碳材料在合成和应用方面面临的问题和挑战进行了总结,并对这类碳材料未来的发展趋势进行了展望。
作为绿色电化学能源的典型代表,二次电池在人们的日常生活中起着越来越重要的作用,其中锂离子电池是目前市场上较为常用的储能器件,具有高能量密度、高工作电压、低自放电及环境友好等诸多优点[41]。但锂离子电池仍旧存在一些尚待解决的问题,比如安全性和金属锂的高成本问题[42],以及现有容量已不能满足需求等,因此,人们将目光投向了具有更高能量密度的锂硫电池及储量丰富且成本低的钠离子电池。在所有这些二次电池中,电极材料的作用最为关键,发展高容量、长寿命、绿色环保的高性能电极材料是二次电池目前的研究热点。MOFs衍生的中空碳材料的大比表面积、多孔结构及内部空腔,可极大地改善电解液的浸润性,作为电极材料在锂离子电池中能够提供比石墨更高的容量,在锂硫/硒电池中可提高活性硫或硒的负载量,在钠离子电池中显示出更有利的钠离子传输和扩散[43-44]。此外,碳材料内部均匀掺杂的杂原子又可进一步提升材料的电化学活性[45]。
高能量密度的电极材料一直是研究人员追寻的目标。具有较高理论比容量的硅、锗等已被尝试用来替换商业的石墨负极材料,但这类材料在充放电反应过程中巨大的体积变化极易导致电极材料的粉化剥落,严重阻碍了其商业化的应用[46]。相比之下,碳基材料结构稳定,导电性好,通过合理构建微纳结构可提升其电化学活性,进而提高比容量。MOFs衍生的中空碳材料利用其独特的多孔及内部中空结构,可有效地缩短锂离子的扩散路径,增大电极材料与电解液的有效接触面积,而其较高的比表面积及较低的质量密度也为比容量提升奠定基础[47]。
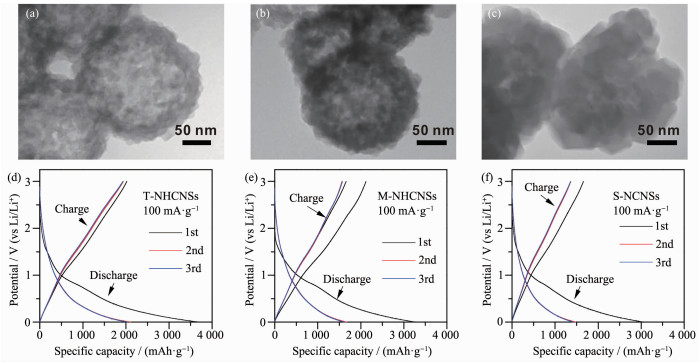
Yang等[48]首先采用乳液基界面反应合成ZIF-8纳米球,然后在不同温度下碳化制备具有多壳层厚度及不同氮掺杂含量的多孔中空碳材料(NHCNSs)(图 2)。该材料具有1 038 m2·g-1的大比表面积及仅几十纳米厚的多孔壳层,可提供较大的电化学活性面积及为电解质离子提供较短的传输路径。同时较高的石墨化程度明显增强材料的导电性,高吡啶氮和吡咯氮掺杂有效提升了材料的比容量,因此NHCNSs在作为锂离子电池负极材料时,显示出了优异的电化学性能。当壳层厚度为20 nm,氮含量为16.6%(n/n)时,该材料在100 mA·g-1的电流密度下具有2 053 mAh·g-1的比容量,且在5.0 A·g-1下循环1 000圈后,材料的比容量仍可达到879 mAh· g-1。此外,若基于负极材料的质量计算,NHCNSs在能量密度为500 Wh·kg-1时,具有50 W·kg-1的功率密度,在1 650 W·kg-1的高功率密度下依然保持有290 Wh·kg-1的高能量密度。
锂硫电池是一种以金属锂为负极,硫单质为正极的二次电池,其具有较高的理论比容量(1 675 mAh·g-1),能量密度更是高达2 600 Wh·kg-1。单质硫对环境友好,在地球上储量丰富,且价格相对低廉,促使锂硫电池成为最具发展前景的下一代二次电池之一[49]。但锂硫电池在实际应用中很难达到其理论容量,这主要是因为硫单质的导电性差(5×10-30 S· m-1),而且硫正极在电化学反应中生成的长链多硫化物(Li2Sx,4≤x≤8)易溶解在电解液中,并在电池正负极之间穿梭,致使反应中生成的绝缘性短链Li2S2或Li2S沉积在电极表面,造成活性物质的流失及较差的循环性能[50]。除此之外,硫单质在电化学嵌锂及脱锂反应中发生巨大的体积形变,极易导致电极材料的粉化及脱落,进一步导致电极结构坍塌,电池寿命变短[51]。解决这些问题最有效的方法一般是将活性硫限域在导电载体材料中,提高电极导电性的同时,抑制多硫化物的穿梭效应。MOFs衍生的中空多孔碳材料凭借其大比表面、高孔隙率及均匀的杂原子掺杂成为了限域活性硫的极佳宿主载体[52]。
本课题组[38]利用单宁酸对ZIF-8前驱体在水溶液中进行温和蚀刻,先制得空心型的ZIF-8多面体,然后经惰性气氛碳化后成功制备了具有多级孔壳层结构的中空多面体型氮掺杂碳材料(HNC),并将此材料作为硫正极的载体(S@HNC)(图 3)。相比ZIF-8衍生的微孔实心型氮掺杂碳载体(NC),空心型HNC载体同时具有微孔、介孔及较大的内部空腔,便于大尺寸的S8分子快速浸入载体材料内部,实现高的活性硫载量。这种氮掺杂的多级孔材料可对链状多硫化物进行快速吸附,如对Li2S6分子显示出快速的吸附行为(10 min内)。得益于其独特的结构,空心型HNC载体在72%(w/w)的高载硫量下仍具有较好的倍率性能及循环稳定性,且比实心型NC载体材料显示出更高的容量、更优的倍率性能及更好的循环稳定性。

比起单纯的氮掺杂,多种杂原子掺杂的中空碳材料作为硫正极的载体时显示出更优异的性能。例如,Zhang等[53]采用双溶剂法,将不同质量的硫氰酸铵(AT)浸入NH2-MIL-101(Al)内部,经600 ℃热处理及酸洗除去金属铝后,制得了包含g-C3N4纳米点的氮、硫共掺杂中空碳材料(CN@NSHPC)。NH2-MIL-101(Al)的孔结构对g-C3N4纳米点的生长具有一定的限域作用,限制其颗粒尺寸小于5 nm。多种组分间的协同作用促使CN@NSHPC展现出比热解单纯NH2-MIL-101(Al)或AT得到的氮掺杂多孔碳(NPC)或g-C3N4更优的多硫化物吸附性能。在负载活性硫后,制得的S/CN@NSHPC电极在0.2C(1.0C=1 672 mA·g-1)的电流密度下具有1 447 mAh·g-1的比容量,在5.0C的电流密度下的倍率比容量可达387 mAh·g-1,且在1.0C下循环500圈后,每圈的比容量损失仅为0.048%。
除了仅利用MOFs来制备中空多孔碳材料,MOFs与其他种类前驱体的复合物也可用来制备具有多孔结构的中空碳材料。Li等[39]选取离子型咪唑鎓基有机聚合物包覆的ZIF-8复合物为前驱体,将有机聚合物中的溴离子交换为二氰胺。依据有机聚合物的低热稳定性及ZIF-8较差的化学和水稳定性,在热解过程中,有机聚合物先分解,其释放的易挥发分子被内部的ZIF-8吸附,浸入到ZIF-8孔道内,并进一步对ZIF-8进行蚀刻,随着热解过程的不断持续,最终制得中空的多孔氮掺杂碳材料(HNPC)。HNPC材料具有超好的稳定性及多级孔特性,其中空结构及高掺杂氮量对多硫化物的吸附及传质过程都极为有利,而完美的融合结构又可为电子在整个电极的快速传输提供保证,因此HNPC材料在锂硫电池中展现出优异的整体性能。在900 ℃热解得到的HNPC-900,经负载65%(w/w)的硫单质后,HNPC-900-65S正极在2.0C(1.0C=1 675 mA·g-1)的电流密度下循环800圈后,其比容量仍有562 mAh·g-1,平均每圈的容量损失率低至0.035%。
硒元素与硫共属同一主族,因而二者的化学性质相似,具有类似的电化学嵌锂及脱锂反应机制,其理论质量比容量为678 mAh·g-1,理论体积比容量达到3 253 mAh·cm-3 [54]。相比于硫,硒具有更高的导电性(1×10-3 S·m-1),可实现快速的电化学反应过程[55]。但与锂硫电池相似,硒在电化学反应过程中也存在大体积膨胀及反应中间物在电解质中溶解的问题,这些问题导致锂硒电池中的活性物利用率较低、循环稳定性及库伦效率较差等不良现象[56]。为了缓解上述问题,Park等[57]将ZIF-8的纳米颗粒分散到聚丙烯腈(PAN)的溶液中,通过静电纺丝法制得ZIF-8/PAN纳米纤维(ZIF-8/PAN NF),经碳化及酸洗除去金属锌后,ZIF-8转变为中空的多面体型碳,而PAN则变成带有介孔结构的碳纤维。上述二者的复合产物M-CNF通过KOH在800 ℃活化,制得兼具微孔及介孔结构的双重孔材料BP-CNF。将BP-CNF材料与硒单质复合,最终制得BP-CNF/Se正极材料(图 4)。在该正极材料中,介孔结构保证材料具有较好的电解液浸润性,而大量的微孔可提高材料中短链硒化物的利用率,故而BP-CNF/Se材料作为锂硒电池正极时,显示出较高的放电比容量及优越的倍率性能。在0.5C(1.0C=675 mA·g-1)的电流密度下,BP-CNF/Se正极的第2圈及第300圈放电比容量分别为742和588 mAh·g-1,相当于79.2%的容量保持率,且在10.0C的高倍率下,仍有568 mAh·g-1的放电比容量。

Liu等[58]同样以ZIF-8为前驱体,热处理得到了具有中空内核、介孔内壳层及微孔外壳层的立方体型碳材料(HMCNCs)。他们首先在ZIF-8的立方体型纳米颗粒外部包覆不同厚度的介孔二氧化硅(m-SiO2),然后对此复合材料进行碳化处理。因为碳化优先在m-SiO2层与ZIF-8颗粒的界面处进行,而较高厚度m-SiO2层在碳化的过程中可以抵消核壳材料的向内收缩,故内部的ZIF-8在热解过程中向外收缩,诱使复合材料在除去m-SiO2后,最终生成具有介微孔壳层的中空HMCNCs材料。该材料的孔尺寸可达25 nm,具有1 086 m2·g-1及3.77 cm3·g-1的大比表面积和孔容。在负载SeS2后,SeS2/HMCNCs正极材料在0.2 A·g-1的电流密度下循环100圈后,其比容量可维持在812.6 mAh·g-1,在5.0 A·g-1的高电流密度下,仍具有455.1 mAh·g-1的倍率比容量。
由于锂资源有限,在地球上储量不多且分布不均,这势必会大大增加其器件成本,限制其更大范围的应用。而与锂同族的钠元素在地球上储量丰富,分布广泛,提取成本较为低廉。同时,钠元素具有与锂相似的物理和化学性质,因此钠离子电池在储能机理及结构组成上均与锂离子电池类似,故而钠离子电池在未来将有望取代锂离子电池,成为可大规模使用的一种储能装置[59]。然而,尽管两种电池的工作原理基本相似,锂离子电池的一些电极材料也可作为钠离子电池的关键材料,但由于钠离子的半径及原子量均比锂离子大很多,严重影响了钠离子在电化学反应中的迁移,导致钠离子不能在石墨材料中可逆脱嵌,使得石墨无法作为钠离子电池的负极材料[60]。因此,开发高性能的可储钠负极材料成为实现钠离子电池发展和应用的关键因素之一。
Zhang等[61]以ZIF-8纳米晶为前体,经化学蚀刻得到壳层厚度为10 nm的气泡型ZIF-8前驱体。该气泡型ZIF-8延续了纳米晶的多面体形貌,颗粒尺寸在100 nm左右,且仍保留了单晶特性。将此前驱体在惰性气氛下碳化及酸洗除去金属锌或氧化锌后,可制得高比表面(比表面积为700 m2·g-1)的中空氮掺杂多孔碳材料。制得的碳材料具有大量的微孔、介孔以及40 nm左右的空腔。作为钠离子电池的负极材料,中空型碳材料比未经蚀刻的实心型碳展现出更优的倍率和循环性能。在10.0 A·g-1的电流密度下循环1 000次后,中空型碳电极的比容量没有任何衰减,可一直保持~99%的库仑效率。
Chen等[62]采用静电纺丝技术先制得醋酸锌、醋酸钴与PAN的复合纤维纸,再将此复合纤维纸浸泡于含有2-甲基咪唑的乙醇溶液中,通过金属与配体的配位作用,在PAN纤维上生长双金属咪唑框架聚合物(BMZIF)层,得到具有核-壳结构的纤维状复合物PAN/Zn(Ac)2/Co(Ac)2@BMZIF。进一步将该复合物在700 ℃碳化,BMZIF层中ZIF-8转变为高含氮量的多孔碳,而ZIF-67在碳化后生成高石墨化的碳材料。此外,Zn(Ac)2降解生成的ZnO对内部的PAN纤维产生蚀刻作用(ZnO+C → Zn+CO2/CO),在酸洗除去金属后,成功制得中空型高石墨化氮掺杂多孔碳纤维(CHTs)。该碳纤维仍具有较好的韧性和柔性,可作为柔性电极材料使用。在不添加导电剂和胶黏剂的情况下,将CHTs直接作为钠离子电池的负极,该负极材料展现出较高的放电比容量和突出的倍率性能,在4.5 A·g-1的高电流密度下循环10 000次后,其比容量(~140 mAh·g-1)也没有明显的下降,显示出极为优异的循环稳定性。最重要的是,在经历10 000次的长循环后,CHTs电极材料仍维持了初始的中空结构,其形貌和结构依然完好,没有发生大的改变。这些优异的性能主要得益于特意设计的中空多孔纤维结构,其中碳材料大的层间距及丰富的缺陷保证充足的储钠位点,高比表面积及多孔结构有助于电解液与活性材料的充分接触,同时缩短钠离子的扩散路径,保证快速的电荷转移反应,而高长径比的管状结构可实现快速的一维电子传输及电渗流。
超级电容器又叫电化学电容器,是介于传统电容器和二次电池之间的一种独特储能器件,兼具传统电容器功率密度高及二次电池能量密度高的储能特性,具有倍率性能佳、安全性能高、循环寿命长及绿色无污染等诸多优点,因此被广泛用于不间断电源、汽车工业等诸多领域,受到学术界和工业界的高度关注。超级电容器的储能方式主要有两种,一种是利用双电层实现电容存储,即通过电解质离子与电极界面形成的电荷分离来存储能量,另一种方式是法拉第赝电容储能,即利用在电极表面或体相发生的可逆氧化还原反应来实现电容存储。目前,超级电容器的性能主要由电极材料决定。在众多的电极材料中,碳材料是使用最为广泛的一种电极材料,是目前市售的超级电容器主要电极材料,主要是因为碳材料来源广、无污染、导电性好、比表面积大等优点[63]。一般情况下,碳材料都是通过电解质离子在其表面的电荷分离形成双电层来储能,所以大比表面积及合适的孔径是实现高电容的关键方法[64]。尽管通过物理或化学活化法可使一些有机前驱体转变为高比表面积的碳材料,但这类碳材料的孔结构往往存在较多缺陷,易导致离子传输的动力学问题,致使材料的倍率性能不佳[65]。研究表明,作为超级电容器的电极材料,高孔隙率及部分石墨化的碳材料可有效提高其比表面积的利用率,降低大倍率极化,进而获得高性能的器件设备。MOFs衍生的碳材料很好地满足了上述要求,以MOFs为前驱体制备的多孔碳材料具有可调的大比表面积及规则的纳米孔结构,为构建高活性的电极材料打下基础[66-68]。不过,以单纯MOFs为前驱体制备的碳材料也存在一些缺点,比如因为大多数MOFs都为微孔结构,由此得到的碳材料也基本保持了前驱体的微孔结构,而这种全微孔结构对电解质离子的快速扩散和渗透造成一定阻碍,导致有效接触面积降低及较差的倍率性能[69]。最近,基于MOFs前驱体制备的中空多孔碳材料受到了人们越来越多的重视,这主要是因为中空多孔碳材料比实心型的微孔碳材料具有更高的可接触面积,便于离子的快速传输,可适应充放电过程中的体积变化,促进电极的反应动力学[70-72]。
ZIF-8因合成简单、形貌可控及高氮含量等优点,经常被选做制备多孔碳材料的前驱体。例如,Li等[73]以电解质修饰的二氧化硅为模板,在此模板上沉积生长ZIF-8,经碳化及除去模板后,制得中空的氮掺杂碳壳层框架。该碳材料作为超级电容器的电极材料时,在6 mol·L-1 KOH电解液中,电流密度为1.0 A·g-1的比电容为253.6 F·g-1,提升电流密度至50.0 A·g-1时,电容保持率仍有79%,且经过20 000次循环后的比电容保持率可达92.1%。此外,由此材料组装的对称型电容器在1 mol·L-1 Na2SO4电解液中,电容器可显示1.6 V的高电压,其能量密度可达到13.3 Wh·kg-1。同样以ZIF-8为前驱体,Wang等[37]先将ZIF-8纳米颗粒(ZIF-8 NPs)分散到PAN的N,N-二甲基甲酰胺(DMF)溶液中,采用静电纺丝法制得ZIF-8与PAN的复合纤维(ZIF-8/PAN),后经高温碳化成功制得一维纳米多孔碳纤维(NPCF)(图 5a)。在碳化过程中,ZIF-8 NPs转变为空心型的立方体壳层结构,并被牢牢束缚在由PAN热解得到的一维碳纤维上。得益于独特的一维中空纤维结构,NPCF展现出比单纯ZIF-8衍生的碳材料更好的电化学性能。如图 5(b,c)所示,NPCF在不同扫速下,其循环伏安曲线均为规则矩形,说明比电容主要来源于电双层电容。不同电流密度下的恒流充放电曲线较小的电压降,表明NPCF具有较好的导电性,因此NPCF作为超级电容器电极材料,电流密度为1.0 A·g-1的比电容有332 F·g-1,且在此电流密度下循环5 000次后,电容保持率为98.9%。随后,Chen等[74]以ZIF-8及PAN为前驱体,同样利用静电纺丝法制备出包含中空纳米颗粒的氮掺杂碳纤维。作为超级电容器电极材料,该纤维电极在1.0及50.0 A·g-1电流密度下的比电容分别为307.2及193.4 F·g-1,在功率密度为25 000 W·kg-1时的最大能量密度可达10.96 Wh·kg-1。该纤维电极同时展现出优异的循环稳定性,在5.0 A·g-1循环10 000次后,比电容的损失率仅为1.8 %。

除了常见的氮原子掺杂,硫、磷等杂原子的掺杂可进一步增加碳材料的亲水性,提高电解液浸润性,同时提供更多的表面法拉第反应位点,进而有效改善碳材料的电化学性能。Zhu等[75]利用双溶剂法将硫脲引入MIL-101-NH2中,在惰性气氛下热解制得中空纳米胶囊状的氮、硫共掺多孔碳材料。该材料具有多级孔(大孔、介孔及微孔)结构,有效增加了可接触面积,利于电荷的快速转移。凭借多级孔结构与多种杂原子掺杂的协同效应,该碳材料表现出优异的电化学性能。作为超级电容器电极材料,在电流密度为10.0 A·g-1时的比电容值为240 F·g-1。当使用离子液体作为电解质时,该材料在功率密度为8 250 W·kg-1时的能量密度可达46.7 Wh·kg-1。Zhang等[76]先在ZIF-8纳米颗粒的表面生长有机聚合物,然后经碳化及酸洗后成功制得中空多面体型的氮、磷、硫共掺的多孔碳材料(ZIF-8@PZS-C)(图 6a和b)。与单纯ZIF-8衍生的碳材料(ZIF-8-C)相比,该法制备的碳材料具有更大的比表面积、更高的导电性及极好的亲水表面。如图 6(c,d)所示,无论在多大的扫速下,ZIF-8@PZS-C材料均比ZIF-8-C表现出更高的比电容值,说明ZIF-8@PZS-C具有更好的双电层电容性能。即使在10.0 A·g-1的电流密度下循环10 000次后,ZIF-8@PZS-C的恒流充放电曲线与初始几周的曲线仍基本一致,表明该材料突出的循环稳定性。这些优异的性能主要来源于其特殊结构与多种杂原子掺杂的协同作用。

氧还原反应是燃料电池和金属空气电池中的一个关键反应过程,其效率直接影响了电池的转化效率,因此探寻高效的氧还原电催化剂来克服氧气缓慢的反应动力学问题是提升这类能量转换装置效率的关键。虽然Pt/C催化剂在目前的氧还原反应中表现出较高的活性,但其较差的稳定性及高昂的价格严重阻碍了商业化的大规模应用[77]。近来,价格低廉且资源丰富的纳米碳催化剂引起了越来越多的关注,主要是因为这类碳基催化剂在氧还原反应中展现出较好的抗一氧化碳和甲醇活性,并具有氧还原催化的四电子反应过程。在这类碳材料中,基于MOFs前驱体制备的中空碳材料更是显示出可与Pt/C催化剂相媲美的氧还原催化活性,其高催化活性可归因于高的比表面积和孔隙率,以及有序的中空结构暴露的更多催化活性位点,保证催化位点与电解液的充分接触,进而实现材料的高催化性能[78-79]。此外,MOFs衍生的碳材料中均匀的杂原子掺杂可改变周围碳原子的表面电子状态,增强氧气吸附的同时促进氧气的活化解离,从而进一步提高此类碳材料的氧还原催化活性[80-81]。
Zhang等[40]先在低温下碳化MOF-5,然后将除去氧化锌后得到的多孔碳与甲酸镍复合,在1 000 ℃的高温下利用金属镍催化多孔碳生成石墨化多孔碳(GPC)。之后水热除去金属镍颗粒,将GPC与尿素混合,在不同温度下碳化,最终制得氮掺杂石墨化多孔碳与碳纳米管的复合材料(NGPC/NCNTs)(图 7a,b)。得益于丰富的氮掺杂、高的石墨化程度及大的比表面积,该碳材料在氧还原反应中表现出非常高的电催化活性。其中,900 ℃下制得的NGPC/NCNT-900材料具有比Pt/C更好的活性、更优的稳定性和抗甲醇性能(图 7(c~e))。Chai等[82]以均苯三甲酸和硝酸锌构筑的配位聚合物微球为初始前驱体,在空气氛下热解得到氧化锌微球,然后以此微球为金属源及模板生长ZIF-8,制得ZnO@ZIF-8核壳型微米颗粒,并以此核壳微米颗粒为前驱体在1 000 ℃碳化制得中空的氮掺杂碳球(HNCSs)。作为氧还原的电催化剂,HNCSs的扩散极限电流可达5.34 mA·cm-2,Tafel斜率为65.7 mV·dec-1,与Pt/C (62.6 mV·dec-1)基本接近。恒压下的计时电流曲线显示HNCSs催化剂具有较好的电催化稳定性,经过10 h的测试,电流保持率仍有96.5%。Wu等[83]同样以ZIF-8为前驱体,先在ZIF-8表面包覆一层单宁酸(TA),形成ZIF-8@TA核壳复合物,然后利用1,4-苯二硼酸与TA之间的相互作用,引入硼源,将此前驱体碳化后制得氮、硼共掺杂的中空多孔碳材料。该碳材料具有936 m2·g-1的高比表面积,在氧还原测试中显示出-0.12 V(vs Ag/AgCl)的起始电位,具有突出的氧还原活性、优异的抗甲醇性能及催化稳定性。第一性原理计算表明,氮、硼共掺杂的协同效应可产生更多的活性位点,进而降低决速步的自由能变值,提高其催化活性。

多孔碳材料因比表面积大、原料来源广、稳定性高及对环境无污染的等特性被广泛地用于储能器件及电催化氧还原反应中。相比于常见的由有机分子或生物质材料制备的多孔碳材料,以MOFs为前驱体,通过在惰性气氛下高温热解并除去金属后得到的多孔碳材料,一般不需要再经过后续复杂的物理或化学活化,即可得到具有大比表面积及高孔隙率的碳材料,且这类材料通常保留了MOFs前驱体规整的网络孔道结构及特有的形貌,同时MOFs配体中的杂原子又可为这类碳材料提供丰富、均匀的杂原子掺杂。此外,因为MOFs本身结构多种多样,且组成丰富,由此可以选用不同类型的MOFs前驱体来制备孔道结构多样、多种杂原子掺杂及不同比表面积的多孔碳材料,并以此实现对所得多孔碳材料结构及性能的调控。在此基础上,MOFs衍生的中空结构的多孔碳材料展现出更多的结构优势。与实心型的多孔碳相比,中空结构的多孔碳具有更好的电解液浸润性、更高的有效电化学接触面积、更短的离子扩散与电子传输距离、及可容纳充放电过程中更大的体积膨胀等,故而中空结构的多孔碳可实现高传质速率,往往具有突出的大倍率性能,进而表现出优异的电化学活性。基于此,本文介绍了近年来MOFs衍生的中空多孔碳材料的相关研究进展,主要包括这类碳材料的制备及其在二次电池、超级电容器及电催化氧还原等领域的应用。尽管由MOFs制备的中空碳材料因其独特的结构优势成为近年来的研究热点,且取得了一定的研究进展,但其未来的发展仍面临许多的问题与挑战:
(1) 虽然MOFs有成千上万种,但目前被用作前驱体的多数还是一些经典结构的MOFs,如ZIF或MIL系列等。有限的前驱体选择导致得到的碳材料往往表现出相似的形貌及孔结构,不利于丰富此类材料的结构类型及进一步改善与提升性能。因此,未来仍需不断探索与发展其他结构与组成的MOFs作为此类碳材料的前驱体,以期得到性能更为优异的中空多孔碳材料。
(2) MOFs包含金属离子与有机配体2种组分,在高温碳化后,有机配体转变为碳材料,而金属也被保留下来,并与碳材料形成复合物。尽管含有金属Zn的MOFs在热解过程中,Zn可以通过高温挥发除去,但大部分MOFs在碳化后仍需要经过酸或碱处理除去金属后才可得到碳材料,这就使得制备过程更加繁琐,且高浓度的酸碱使用也具有一定的危险性。此外,有机配体中的杂原子在高温碳化过程中大量损失,使得掺杂含量较低。开发过程温和及最大限度保留杂原子的制备条件是目前急需解决的问题。
(3) 与传统方法制备的多孔碳材料相比,以MOFs为前驱体制备的多孔碳材料显现出更多的结构和性能优势,但同时也存在致命的缺点,就是其成本高昂。虽然一些经典MOFs可以选用廉价的金属离子和配体,但其较低的产率又往往使得造价不断上涨,故由MOFs衍生的碳材料的进一步发展需要克服其产率低的难题,同时需要开发廉价的有机配体以实现大规模的生产。
(4) 通过模板法或预先蚀刻MOFs前驱体法均可制得中空多孔碳材料。除此之外,MOFs通过热解碳化及后续除去金属后,也可得到中空多孔碳材料。但上述方法得到材料的结构或取决于模板或存在很大程度的结构不确定性,致使最终材料的组成及结构难以控制,对研究其构效关系造成极大的障碍。因此,仍需要进一步深入探究MOFs前驱体结构以及其在碳化过程中的结构衍变,从而实现最终碳材料的结构控制,以满足实际需求。
总之,以MOFs为前驱体制备的中空碳材料具有比传统方法制备的碳材料更多的结构优势,且在二次电池、超级电容器及电催化氧还原等领域显示出更为优异的电化学性能。相信随着研究的不断深入,目前存在的一些问题将会逐步得到改善和解决,并促进该研究领域的进一步发展,为MOFs的应用及碳材料的开发和实用化开辟新的方向和路径。
Li S, Niu J J, Zhao Y C, et al. Nat. Commun., 2015, 6:7872 doi: 10.1038/ncomms8872
Stern P C, Sovacool B K, Dietz T. Nat. Clim. Change, 2016, 6:547-555 doi: 10.1038/nclimate3027
Quadrelli R, Peterson S. Energy Policy, 2007, 35:5938-5952 doi: 10.1016/j.enpol.2007.07.001
Xia W, Mahmood A, Zou R Q, et al. Energy Environ. Sci., 2015, 8:1837-1866 doi: 10.1039/C5EE00762C
Long W Y, Fang B Z, Ignaszak A, et al. Chem. Soc. Rev., 2017, 46:7176-7190 doi: 10.1039/C6CS00639F
Service R F. Science, 2006, 313:902 doi: 10.1126/science.313.5789.902
Zhang J T, Zhao Z H, Xia Z H, et al. Nat. Nanotechnol., 2015, 10:444-452 doi: 10.1038/nnano.2015.48
Cavaliere S, Subianto S, Savych I, et al. Energy Environ. Sci., 2011, 4:4761-4785 doi: 10.1039/c1ee02201f
Etacheri V, Marom R, Elazari R, et al. Energy Environ. Sci., 2011, 4:3243-3262 doi: 10.1039/c1ee01598b
Zhang W, Zhu S Y, Luque R, et al. Chem. Soc. Rev., 2016, 45:715-752 doi: 10.1039/C5CS00297D
Salunkhe R R, Kaneti Y V, Kim J, et al. Acc. Chem. Res., 2016, 49:2796-2806 doi: 10.1021/acs.accounts.6b00460
Xu B, Zheng D F, Jia M Q, et al. Electrochim. Acta, 2013, 98:176-182 doi: 10.1016/j.electacta.2013.03.053
Xiao P W, Meng Q H, Zhao L, et al. Mater. Des., 2017, 129:164-172 doi: 10.1016/j.matdes.2017.05.035
Chen L F, Huang Z H, Liang H W, et al. Adv. Funct. Mater., 2014, 24:5104-5111 doi: 10.1002/adfm.201400590
Jiang L, Yan J W, Hao L X, et al. Carbon, 2013, 56:146-154 doi: 10.1016/j.carbon.2012.12.085
Furukawa H, Cordova K E, O'Keeffe M, et al. Science, 2013, 341:1230444 doi: 10.1126/science.1230444
Lu W G, Wei Z W, Gu Z Y, et al. Chem. Soc. Rev., 2014, 43:5561-5593 doi: 10.1039/C4CS00003J
Liu B, Shioyama H, Akita T, et al. J. Am. Chem. Soc., 2008, 130:5390-5391 doi: 10.1021/ja7106146
Hu M, Reboul J, Furukawa S, et al. J. Am. Chem. Soc., 2012, 134:2864-2867 doi: 10.1021/ja208940u
Yang S J, Kim T, Im J H, et al. Chem. Mater., 2012, 24:464-470 doi: 10.1021/cm202554j
Zheng F C, Yang Y, Chen Q W. Nat. Commun., 2014, 5:5261 doi: 10.1038/ncomms6261
Zhang L J, Su Z X, Jiang F L, et al. Nanoscale, 2014, 6:6590-6602 doi: 10.1039/C4NR00348A
Li J S, Li S L, Tang Y J, et al. Sci. Rep., 2014, 4:5130-5137
Song Z X, Liu W W, Cheng N C, et al. Mater. Horiz., 2017, 4:900-907 doi: 10.1039/C7MH00244K
Liu J L, Zhu D D, Guo C X, et al. Adv. Energy Mater., 2017, 7:1700518 doi: 10.1002/aenm.201700518
Kaneti Y V, Tang J, Salunkhe R R, et al. Adv. Mater., 2017, 29:1604898 doi: 10.1002/adma.201604898
Guan B Y, Yu X Y, Wu H B, et al. Adv. Mater., 2017, 29:1703614 doi: 10.1002/adma.201703614
Yang W P, Li X X, Li Y, et al. Adv. Mater., 2018, 31:1804740
Wang L, Han Y Z, Feng X, et al. Coord. Chem. Rev., 2016, 307:361-381 doi: 10.1016/j.ccr.2015.09.002
Xie X C, Huang K J, Wu X. J. Mater. Chem. A, 2018, 6:6754-6771 doi: 10.1039/C8TA00612A
Cai Z X, Wang Z L, Kim J, et al. Adv. Mater., 2019, 31:1804903 doi: 10.1002/adma.201804903
Wang C H, Kaneti Y V, Bando Y, et al. Mater. Horiz., 2018, 5:394-407 doi: 10.1039/C8MH00133B
Yang S L, Peng L, Huang P P, et al. Angew. Chem. Int. Ed., 2016, 55:4016-4020 doi: 10.1002/anie.201600455
Hu X R, Wang C H, Li J S, et al. ACS Appl. Mater. Interfaces, 2018, 10:15051-15057 doi: 10.1021/acsami.8b02281
Lee H J, Choi S, Oh M. Chem. Commun., 2014, 50:4492-4495 doi: 10.1039/c4cc00943f
Shen K, Chen X D, Chen J Y, et al. ACS Catal., 2016, 6:5887-5903 doi: 10.1021/acscatal.6b01222
Wang C H, Liu C, Li J S, et al. Chem. Commun., 2017, 53:1751-1754 doi: 10.1039/C6CC09832K
Yang D H, Zhou H Y, Liu H, et al. iScience, 2019, 13:243-253 doi: 10.1016/j.isci.2019.02.019
Li Z L, Xiao Z B, Wang S Q, et al. Adv. Funct. Mater., 2019, DOI: 10.1002/adfm.201902322.
Zhang L J, Wang X Y, Wang R H, et al. Chem. Mater., 2015, 27:7610-7618 doi: 10.1021/acs.chemmater.5b02708
Dubal D P, Ayyad O, Ruiz V, et al. Chem. Soc. Rev., 2015, 44:1777-1790 doi: 10.1039/C4CS00266K
Tarascon J M, Armand M. Nature, 2001, 414:359-367 doi: 10.1038/35104644
Li X X, Zheng S S, Jin L, et al. Adv. Energy Mater., 2018, 8:1800716 doi: 10.1002/aenm.201800716
Zhong M, Kong L J, Li N, et al. Coord. Chem. Rev., 2019, 388:172-201 doi: 10.1016/j.ccr.2019.02.029
叶绍凤, 刘文贤, 徐喜连, 等.材料导报, 2018, 32:2129-2142 doi: 10.11896/j.issn.1005-023X.2018.13.001YE Shao-Feng, LIU Wen-Xian, XU Xi-Lian, et al. Mater. Rep., 2018, 32:2129-2142 doi: 10.11896/j.issn.1005-023X.2018.13.001
Zhang W J. J. Power Sources, 2011, 196:13-24 doi: 10.1016/j.jpowsour.2010.07.020
Zhang L, Liu H W, Shi W, et al. Coord. Chem. Rev., 2019, 388:293-309 doi: 10.1016/j.ccr.2019.02.030
Yang Y F, Jin S, Zhang Z, et al. ACS Appl. Mater. Interfaces, 2017, 9:14180-14186 doi: 10.1021/acsami.6b14840
Manthiram A, Fu Y Z, Su Y S. Acc. Chem. Res., 2012, 46:1125-1134
Yang C P, Yin Y X, Guo Y G. J. Phys. Chem. Lett., 2015, 6:256-266 doi: 10.1021/jz502405h
Morozan A, Jaouen F. Energy Environ. Sci., 2012, 5:9269-9290 doi: 10.1039/c2ee22989g
Zheng Y, Zheng S S, Xue H G, et al. J. Mater. Chem. A, 2019, 7:3469-3491 doi: 10.1039/C8TA11075A
Zhang H, Zhao Z B, Hou Y N, et al. J. Mater. Chem. A, 2018, 6:7133-7141 doi: 10.1039/C8TA00529J
Yang C P, Xin S, Yin Y X, et al. Angew. Chem. Int. Ed., 2013, 52:8363-8367 doi: 10.1002/anie.201303147
Luo C, Xu Y H, Zhu Y J, et al. ACS Nano, 2013, 7:8003-8010 doi: 10.1021/nn403108w
Luo C, Zhu Y J, Wen Y, et al. Adv. Funct. Mater., 2014, 24:4082-4089 doi: 10.1002/adfm.201303909
Park S K, Park J S, Kang Y C. J. Mater. Chem. A, 2018, 6:1028-1036 doi: 10.1039/C7TA09676C
Liu C, Huang X D, Wang J, et al. Adv. Funct. Mater., 2018, 28:1705253 doi: 10.1002/adfm.201705253
Hwang J Y, Myung S T, Sun Y K. Chem. Soc. Rev., 2017, 46:3529-3614 doi: 10.1039/C6CS00776G
Wen Y, He K, Zhu Y J, et al. Nat. Commun., 2014, 5:4033 doi: 10.1038/ncomms5033
Zhang W, Jiang X F, Zhao Y Y, et al. Chem. Sci., 2017, 8:3538-3546 doi: 10.1039/C6SC04903F
Chen Y M, Li X Y, Park K, et al. Chem, 2017, 3:152-163 doi: 10.1016/j.chempr.2017.05.021
Inagakia M, Konno H, Tanaike O. J. Power Sources, 2010, 195:7880-7903 doi: 10.1016/j.jpowsour.2010.06.036
Chmiola J, Yushin G, Gogotsi Y, et al. Science, 2006, 313:1760-1763 doi: 10.1126/science.1132195
Wang D W, Li F, Liu M, et al. Angew. Chem. Int. Ed., 2008, 47:373-376 doi: 10.1002/anie.200702721
Salunkhe R R, Kaneti Y V, Kim J, et al. Acc. Chem. Res., 2016, 49:2796-2806 doi: 10.1021/acs.accounts.6b00460
Zheng S S, Xue H G, Pang H. Coord. Chem. Rev., 2018, 373:2-21 doi: 10.1016/j.ccr.2017.07.002
Zhao Y J, Liu J Z, Horn M, et al. Sci. China Mater., 2018, 61:159-184 doi: 10.1007/s40843-017-9153-x
Gong Y J, Chen R Y, Xu H, et al. Nanoscale, 2019, 11:2492-2500 doi: 10.1039/C8NR09454C
Wang J, Luo X L, Young C, et al. Chem. Mater., 2018, 30:4401-4408 doi: 10.1021/acs.chemmater.8b01792
Klosea M, Reinhold R, Pinkert K, et al. Carbon, 2016, 106:306-313 doi: 10.1016/j.carbon.2016.05.046
Xin L J, Chen R R, Liu Q, et al. New J. Chem., 2017, 41:12835-12842 doi: 10.1039/C7NJ02427D
Li Z W, Mi H Y, Liu L, et al. Carbon, 2018, 136:176-186 doi: 10.1016/j.carbon.2018.04.075
Chen L F, Lu Y, Yu L, et al. Energy Environ. Sci., 2017, 10:1777-1783 doi: 10.1039/C7EE00488E
Zhu Q L, Pachfule P, Strubel P, et al. Energy Storage Mater., 2018, 13:72-79 doi: 10.1016/j.ensm.2017.12.027
Zhang J, Fang J H, Han J L, et al. J. Mater. Chem. A, 2018, 6:15245-15252 doi: 10.1039/C8TA04813D
Debe M K. Nature, 2012, 486:43-51 doi: 10.1038/nature11115
Mahmood A, Guo W H, Tabassum H, et al. Adv. Energy Mater., 2016, 6:1600423 doi: 10.1002/aenm.201600423
朱静怡, 梁风, 姚耀春, 等.稀土金属, 2019, 43:186-200ZHU Jing-Yi, LIANG Feng, YAO Yao-Chun, et al. Rare Metals, 2019, 43:186-200
Huang P M, Li H D, Huang X Y, et al. ACS Appl. Mater. Interfaces, 2017, 9:21083-21088 doi: 10.1021/acsami.7b06427
Song Z X, Liu W W, Cheng N C, et al. Mater. Horiz., 2017, 4:900-907 doi: 10.1039/C7MH00244K
Chai L L, Zhang L J, Wang X, et al. Carbon, 2019, 146:248-256 doi: 10.1016/j.carbon.2019.02.006
Wu M C, Li C L, Zhao J, et al. Dalton Trans., 2018, 47:7812-7818 doi: 10.1039/C8DT01517A

图 2 (a~c)薄壳层(T-NHCNSs)、中壳层(M-NHCNSs)及实心型(S-NCNSs)氮掺杂碳材料的透射电子显微镜(TEM)图; (d~f) T-NHCNSs、M-NHCNSs及S-NCNSs材料在100 mA·g-1电流密度下的充放电曲线图[48]
Figure 2 (a~c) Transmission electron microscopy (TEM) images of thin-shell NHCNSs (T-NHCNSs), medium-shell NHCNSs (M-NHCNSs), and solid NCNSs (S-NCNSs); (d~f) Corresponding galvanostatic charge/discharge profiles of T-NHCNSs, M-NHCNSs, and S-NCNSs at the current density of 100 mA·g-1 [48]




图 6 (a) ZIF-8@PZS-C材料的制备示意图; (b) ZIF-8@PZS-C的TEM图; (c) ZIF-8@PZS-C及ZIF-8-C在不同扫速下的比电容; (d) ZIF-8@PZS-C的恒流充放电曲线[76]
Figure 6 (a) Schematic illustration of the preparation of ZIF-8@PZS-C; (b) TEM image of ZIF-8@PZS-C; (c) Specific capacitance of ZIF-8@PZS-C and ZIF-8-C at different scan rates; (d) Galvanostatic charge-discharge curves of ZIF-8@PZS-C[76]

图 7 (a) NGPC/NCNTs材料的制备示意图; (b) NGPC/NCNT-900的TEM图; (c~e) NGPC/NCNT-900和Pt/C的循环伏安曲线、时间-电流密度曲线及加入甲醇后的时间-电流密度曲线[40]
Figure 7 (a) Schematic illustration of the preparation of NGPC/NCNTs; (b) TEM image of NGPC/NCNT-900; (c~e) Cyclic voltammograms, current-time chronoamperometric responses without or with methanol of NGPC/NCNT-900 and Pt/C[40]

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:

 下载:
下载:
 下载:
下载: