
图 1.
(a) CNT@LMR制备流程的SEM图和(b)示意图
Figure 1.
(a) SEM images and (b) schematic illustration of synthesis process of CNT@LMR

随着环境和能源问题的日趋严峻,电能作为清洁能源,已在出行和储能等领域逐步取代化石能源[1]。比如,很多国家开始制定纯电动汽车取代燃油汽车的技术路线图。不过,电动汽车目前还无法取代燃油汽车,这在于其储能装置和动力来源的掣肘,即锂离子电池的低能量和功率密度及循环寿命短的问题[2]。锂离子电池能量密度的提升根本在于其正负极材料比容量密度的提升,因此开发高比容量密度的正负极材料尤为重要[3]。市面上,成熟的锂离子电池正极材料的比容量往往低于200 mAh·g-1,无法使锂离子电池能量密度突破300 Wh·kg-1。富锂锰基正极材料(xLi2MnO3·(1-x)LiMO2,M = Mn,Co,Ni)的比容量可以达到250 mAh·g-1,是实现高能量密度动力电池的优选材料[4]。但是富锂锰基正极材料存在着诸多问题,例如:不可逆容量高和首次库伦效率低(<80%),这不利于锂离子电池电芯的正负极容量(N/P)比的设计;倍率性能差,无法满足动力电池高功率充放电的要求;循环过程中电压衰减严重,导致动力电池的放电电压过低,造成大量的能量损失[5]。面对诸多问题,许多科学家和工程师开展了对富锂锰基正极材料进行很多改进工作[6-7]。
与传统的三元正极材料LiMO2(M = Mn,Co,Ni)不同,富锂锰基正极材料的Li2MnO3相在充电到4.5 V以上会发生电化学反应(Li2MnO3→Li2O+MnO2),这导致富锂锰基正极材料的结构不稳定和电极/电解液的界面反应增多[8-9]。一些研究表明[10-11],对富锂锰基正极材料进行表面改性能够有效稳定其结构和抑制电极/电解液的界面反应。氧化物、氟化物、磷酸盐和碳材料等相继用于对富锂锰基正极材料的表面改性工作[12-14]。碳材料相比于电子惰性化合物材料而言,具有良好的电子电导率和易合成加工性,同时来源广泛且价格低廉[15]。而碳纳米管作为新型的纳米碳材料,具有很多独特优势[16-17]。碳纳米管的一维纤维状结构能够有效地连接电极活性材料,提升材料和电极的电子电导能力;碳纳米管具有高机械强度,添加后能够使电极极片具有较高的韧性,可有效抑制电极材料在充放电循环过程中因体积变化而引起的剥落;碳纳米管具有高长径比和高比表面积,与电极材料均匀复合后,可提高电极材料在电解液中的浸润性。
碳纳米管作为锂离子电池正负极材料的导电添加剂的研究已经较为成熟,将适量的碳纳米管(0.5~5%(w/w))加入电极材料中,能有效提高锂离子电池的功率特性,具有很高的实用价值[18]。但是,作为导电添加剂的碳纳米管在正极材料颗粒间的均匀分散是个难题。通过增加机械分散的强度和时间会破坏正极的二次球形颗粒,加入化学分散剂则会在电极里引入非活性物质。因此,一些工作直接将碳纳米管与正极材料进行复合来解决碳纳米管在材料颗粒间分散难的问题。Guo等[19]将酸化的多壁碳纳米管材料与富锂锰基正极材料复合,在材料表面形成了碳纳米管导电网络,有效地提升了富锂锰基正极材料的倍率性能。不过,富锂锰基正极材料的微观形貌一般是由一次颗粒通过分子间作用力聚集而形成的二次颗粒。碳纳米管与富锂锰基正极材料直接复合,只能在材料的二次球形颗粒表面形成碳纳米管导电网络,虽可以增强二次颗粒之间的电导特性,但却无法改善二次颗粒内部大量的一次颗粒之间的导电状况。
本文提出了碳纳米管改性富锂锰基正极材料的一种新策略。基于课题组前期的工作[20-21],制备了富锂锰基正极材料Li1.184[Ni0.15Mn0.516Co0.15]O2。接着采用气流破碎、高压均质混合分散和喷雾干燥的方法得到了与碳纳米管复合的富锂锰基正极材料,解决了碳纳米管直接作为导电添加剂的均匀分散问题,并实现了碳纳米管在富锂锰基正极材料表面和内部一次颗粒之间均匀分布的效果。通过对改性的材料进行一系列地表征,证实了材料二次球形颗粒表面存在紧密的碳纳米管网状结构,材料内部一次颗粒之间也存在碳纳米管网络。为了评价纳米碳管改性富锂锰基正极的效果,制备了3种电极,分别为碳纳米管改性前、改性后的富锂锰基正极以及用碳纳米管做导电剂的富锂锰基正极,并对其进行了电化学性能测试。对比发现,碳纳米管改性的富锂锰基正极具有最优的倍率性能,证实了碳纳米管导电网络能够大幅度提升富锂锰基正极电导特性。最后,不同循环次数下的电化学阻抗谱表明,分布在富锂锰基正极表面的碳纳米管网状结构改善了电极/电解液界面反应,抑制了电极固体电解质界面(SEI)膜的增厚和电极的过度极化;富锂锰基正极材料颗粒内部的碳纳米管导电网络降低了颗粒间的内阻并加快了电极的电荷转移过程。
富锂锰基正极材料的制备:按化学式Li1.184[Ni0.15Mn0.516Co0.15]O2中各金属元素物质的量之比称取总质量为200 g的MnCO3、NiO、Co3O4、Li2CO3混合粉末,加入1 L去离子水,在球磨机中研磨1 h。然后将混合物浆料转移到砂磨机,再加入1 L去离子水进行超细磨4 h。将超细磨后的浆料进行喷雾干燥,得到喷雾干燥粉末。称取适量粉末置于马弗炉中升温至900 ℃后焙烧20 h,随炉冷却至室温,将获得的粉体过300目筛,得到富锂锰基正极材料(LMR)。
碳纳米管改性富锂锰基正极材料:通过气流破碎机对富锂锰基正极材料进行破碎,二次球形颗粒基本被破碎为小的一次颗粒(LMR broken)。然后加入多壁碳纳米管水分散液(5%(w/w) CNTs,管径为7~11 nm,管长1~10 nm,购自江苏天奈公司),在搅拌式分散机中进行搅拌预分散,富锂锰基正极材料和碳纳米管的质量比为0.99:0.01。将此混合浆料以600 Pa的压力泵入高压均质混合机(Omega®500,德国NETZSCH公司)处理10圈,得到1%(w/w)碳纳米管与富锂锰基一次颗粒复合的浆料(CNT & LMR dispersed)。最后,浆料通过喷雾干燥机进行干燥重新造粒,并在80 ℃烘箱中干燥12h,得到了碳纳米管改性的富锂锰基正极材料(CNT@LMR)。图 1(a)反映了从富锂锰基正极材料破碎到与碳纳米管均匀分散以及重新造粒电镜表征图,图 1(b)示意图形象地展示了这种碳纳米管改性富锂锰基正极材料的方法。
富锂锰基正极制备:将富锂锰基正极材料、导电剂和粘结剂(PVDF)按质量比80:10:10的比例,加入一定量的N-甲基吡咯烷酮(NMP)后在匀浆机中均匀混合,然后通过涂布机将浆料涂覆在铝箔上,充分干燥后可得到富锂锰基正极极片(活性物质面密度为6.0 mg·cm-2)。基于此方法,制备了未经改性的富锂锰基正极(LMR)、经碳纳米管改性的富锂锰基正极(CNT@LMR)和碳纳米管作为导电剂的富锂锰基正极(CNT/LMR)。其中在CNT/LMR电极制备过程中导电剂的添加组分为碳纳米管和导电炭黑的复合导电剂(质量比为0.08:0.92),即碳纳米管的作为导电剂的质量与作为改性材料的质量相同。将富锂锰基正极压实后(压实密度为2.0 g·cm-3),裁切成14 mm的圆片,在真空烘箱中干燥后移入手套箱后组装成扣式半电池。
材料和极片的表观形貌在扫描电子显微镜(SEM)上观测(Hitachi S-4800,日本Hitachi公司),工作电压为10 kV。材料颗粒的结构和形貌在透射电子显微镜(TEM,Tecnai G2 F20,美国FEI公司)观察,管加速电压为200 kV。材料的结构和组成通过X射线衍射(XRD)分析仪(SmartLab,日本Rigaku公司)测试和激光共聚焦显微拉曼散射光谱仪(Raman,日本Horiba公司)上测试分析得到。XRD的测试参数是Cu Kα射线(λ = 0.154 439 nm),管电压为40 kV,管电流为200 mA,扫描范围10°~80°,扫速10°·min-1,步长0.02°。Raman测试的激发波长为532 nm,测试的波长范围是100~3 500 cm-1。扣式半电池的一系列充放电测试在充放电测试仪(武汉蓝电公司)上进行,测试模式设置为倍率充放电(1C = 200 mA·g-1)。扣式半电池的循环伏安(CV)和电化学阻抗谱(EIS)测试在电化学工作站(Autolab 302N,瑞士万通公司)上完成。电化学阻抗谱的交流信号振幅为5 mV,频率为100 kHz~0.1 Hz。电化学交流阻抗谱的等效电路模型和数据拟合在Zview软件上完成。
通过高温固相合成方法得到了富锂锰基正极材料(LMR),通过SEM图可以明显看出该材料是由一次小颗粒团聚形成的二次球形大颗粒材料(图 2(a)),利用高压气流破碎机的高压气流将二次球形颗粒完全打散为一次小颗粒。一次小颗粒与碳纳米管水分散液充分混合并喷雾干燥重新造粒后,能够得到碳纳米管改性的富锂锰基二次球形颗粒(CNT@LMR)。如图 2(b)所示,碳纳米管紧密地缠绕在一次球形颗粒之间,在二次颗粒表面形成了致密的碳纳米管网络。
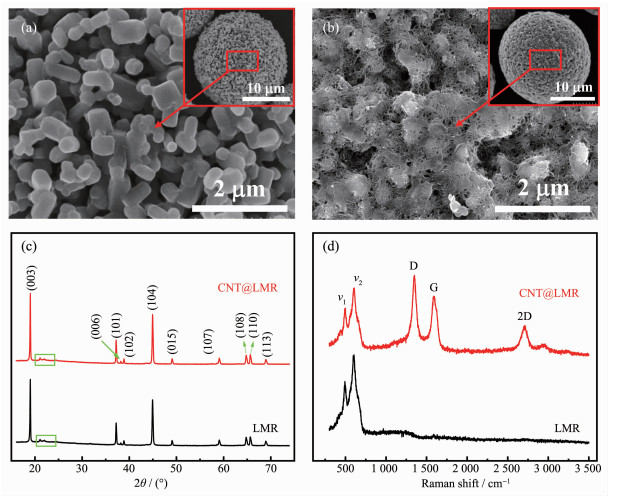
富锂锰基正极材料的XRD图如图 2(c)所示。图中材料的衍射峰尖锐,表明该方法制备的富锂锰基正极材料具有较好的晶体结构。主要衍射峰均与六方α-NaFeO2结构(空间群R3m(166))的衍射峰对应,在20°~25°有一组较小的超晶格峰,这是由于Li2MnO3相过渡金属层中Li和Mn原子的有序排列造成的[22-23]。根据文献报道[24],(003)和(104)晶面衍射峰的强度比是判定Li+和Ni2+之间阳离子混排的指标。本文所制备的富锂锰基正极材料的I(003)/I(104)强度比值为1.33,高于1.2,表明所制备的富锂锰基正极材料的阳离子混排程度较低。同时,从XRD图中可以清楚地观察到(006)/(102)和(108)/(110)晶面衍射峰的分裂,表明该材料具有较好的二维层状结构[25]。CNT@LMR与LMR的XRD图相比,没有发现明显的晶格参数的变化,说明这种机械混合的改性过程温和,碳纳米管没有破坏和影响原始材料的结构。同时,为了更好的确定富锂锰基正极材料表面是否有一层致密的碳纳米管网络结构,通过使用Raman光谱对改性材料进行了测试。如图 2(d)所示,富锂锰基正极材料的拉曼光谱主要是600 cm-1的A1g振动模(ν1)和500 cm-1左右的Eg振动模(ν2)构成,这是由Li2MnO3和LiNi1/3Mn1/3Co1/3O2组分的Raman光谱叠加而成[26-27]。经过碳纳米管改性的富锂锰基材料的拉曼光谱不仅存在450~700 cm-1之间的2个特征峰,同时在1 350 cm-1(G峰)、1 580 cm-1(D峰)和2 700 cm-1(2D峰,也称G′峰)附近出现了碳纳米管的特征峰[28],进而验证了碳纳米管导电网络均匀地分布在富锂锰基正极材料表面。
通过碳纳米管改性富锂锰基正极材料的目的不仅是在富锂锰基正极材料二次颗粒表面形成碳纳米管导电网络,同时要达到碳纳米管穿插在材料内部一次颗粒之间的效果。通过将该复合材料的二次颗粒破碎开后,使用SEM和TEM对材料内部的一次颗粒进行观察。如图 3(a)所示,CNT@LMR材料内部一次颗粒中也有大量的碳纳米管存在。图 3(b)的TEM图表明碳纳米管存在于LMR的一次颗粒之间,其在材料内部的一次颗粒之间构筑的导电网络可以良好地改善材料一次颗粒之间的电导特性。因此,通过二次球形颗粒气流破碎为一次颗粒后,碳纳米管与一次颗粒经过高压均质混合分散,最后喷雾干燥重新造二次球形颗粒的机械物理方法,可以实现碳纳米管在富锂锰基正极材料表面和内部均匀分布的效果。

通过一系列的电化学性能测试来验证碳纳米管改性的富锂锰基复合正极的实用价值。将LMR、CNT@LMR和CNT/LMR的3种正极分别组装成扣式半电池,在电池测试仪上进行相同条件下的对比测试。图 4(a)是富锂锰基3种正极在2.0~4.8 V电压范围内,以0.1C的倍率首次充放电的曲线(1C = 200 mA·g-1)。充电曲线均由4.45 V以下的斜坡区和4.45 V以上的平台区组成,而放电曲线呈现下斜特征。充电4.45 V以下的曲线对应的是LiNi1/3Mn1/3 Co1/3O2组分中锂离子脱出反应。相应地,充电4.45 V以上的平台曲线是Li2MnO3组分电化学活化过程(Li2MnO3→Li2O+MnO2),即锂离子从锂层和金属层脱出,同时伴随着Li2MnO3的晶格氧流失和结构重排[29]。在图 4(a)中,3种富锂锰基正极的充放电曲线的形状和平台均保持一致,说明碳纳米管不论作为改性材料还是导电剂,都未改变富锂锰基材料的本征电化学特性。但是,CNT@LMR具有最高的首次放电比容量256.7 mAh·g-1,且首次库伦效率可达75.6%。而LMR只有240.2 mAh·g-1放电比容量,首次库伦效率也只有73.5%。CNT/LMR的首次放电比容量为248.4 mAh·g-1,高于LMR的首次放电比容量,说明碳纳米管作为导电剂确实改善了富锂锰基正极的电导特性。CNT@LMR具有最高的放电比容量和库伦效率,这是因为碳纳米管在富锂锰基正极材料表面和内部均形成了导电网络,而且其高比表面积增加了电极与电解液的接触面积,这些共同促进了锂离子和电子在充放电过程中在电极表面和内部的迁移。同时,材料表面致密的碳纳米管网络减少了电极与电解液的界面副反应,进而减少了不可逆容量的损失,从而提高了电极的首次库伦效率。
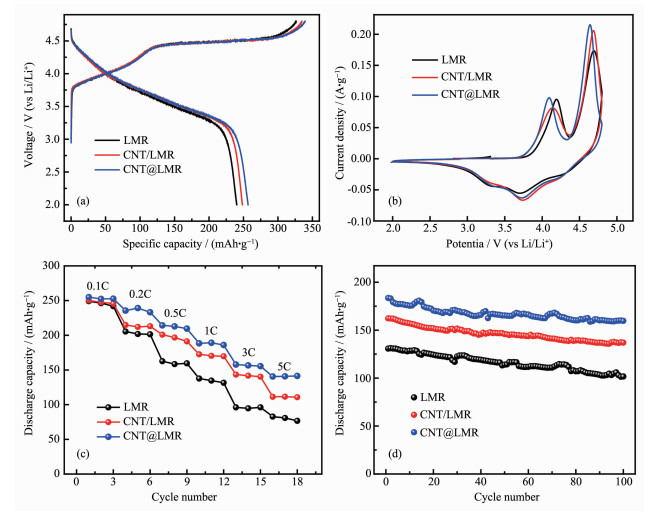
(a) First charge-discharge curves; (b) First CV curves; (c) Rate performance; (d) Cyclic capability performance
图 4(b)显示了3个富锂锰基正极在第1圈的CV曲线,扫描速率设置为0.2 mV·s-1,电压窗口为2.0~4.8 V。富锂锰基正极在4.1 V附近的氧化峰,对应的是Ni2+→Ni4+的氧化反应,而在4.6 V附近的氧化峰,则为Li2MnO3组分中脱锂和不可逆的Li2O生成反应。电极有3个明显的还原峰,分别是在4.22、3.74和3.33 V,对应的是过渡金属离子Co4+、Ni4+和Mn4+的还原[30]。从图 4(b)中看出,相比LMR正极,CNT/LMR和CNT@LMR的氧化峰逐步向左偏移,说明碳纳米管在富锂锰基材料之间形成的导电网络促进了电极中电子的迁移速率,使得过渡金属离子的氧化反应加快[31]。同时,CNT@LMR在4.6 V附近的氧化峰的响应电流强度最高且峰形变窄,表明碳纳米管网络均匀分布在材料表面可以改善高电位下电极/电解液的界面反应,稳定电极材料结构,减少不可逆容量损失,这与图 4(a)中CNT@LMR拥有最高首次库伦效率相一致。
图 4(c)是3种富锂锰基正极的倍率性能对比图。可以清晰地看到,随着电流密度的不断增加,经碳纳米管改性后的富锂锰基正极的高放电比容量优势越发明显。CNT@LMR在5C大电流下的放电比容量为141.4 mAh·g-1,CNT/LMR的放电比容量为110.7 mAh·g-1,而LMR的放电比容量只有76.6 mAh·g-1。在大电流下碳纳米管改性前后的放电比容量相差将近一倍,由此可知,碳纳米管本征的优良电导和比表面积高等特性很好的提高了富锂锰基材料和电极的电导率,提升了电极与电解液的浸润性,从而大幅度提升了富锂锰基正极的倍率性能。
图 4(d)是3种正极在1C倍率下循环100次的循环性能图。CNT@LMR的首次放电比容量为183.6 mAh·g-1,循环100次后放电比容量为160.2 mAh·g-1,容量保持率为87.2%。未经改性的LMR在循环100次后的放电比容量衰减为101.7 mAh·g-1,容量保持率只有77.8%。CNT/LMR在100次后的放电比容量为133.7 mAh·g-1,容量保持率为82.4%。由此可知,碳纳米管改性的富锂正极在1C倍率下循环100次后拥有最高的比容量。这说明了碳纳米管改性的富锂锰基正极不仅能够实现碳管与导电炭黑构成的“点-线”导电网络来促进电极中电子的传导,同时富锂锰基正极材料表面和内部的碳纳米管导电网络,能有效地改善电极/电解液的界面反应,即减缓了过渡金属从材料晶格到电解液中的溶出,抑制了电解液与材料内部活性氧反应后发生的分解。此外,材料表面存在致密的碳纳米管网络层保护了电极材料不被电解液中HF侵蚀,从而在循环次数不断增加的情况下稳定了电极结构[32]。
为了更好地说明碳纳米管改性富锂锰基正极材料倍率性能和循环性能提升的原因,采用SEM和EIS对改性前后的电极进行了表征和测试。图 5为碳纳米管改性富锂锰基正极前后的SEM图,电极极片未经过柱形对辊机辊压,这样可以更好地看清碳管与颗粒之间的形貌。因为导电炭黑为纳米级易于团聚,因此,即使经过匀浆机的快速搅拌导电炭黑仍不能均匀分散在富锂锰基正极材料球形颗粒之间。单纯的导电炭黑只能大幅度改善电极整体的导电性,而无法在颗粒之间构成导电网络(图 5(a,c))。而对于经过碳纳米管改性的富锂锰基正极,碳纳米管不仅弥补了导电炭黑团聚导致的无法充分分散在颗粒之间的不足,起到了连接富锂颗粒的作用,同时与导电炭黑一起协同作用,构筑了“点-线”导电网络。从图 5(b,d)中可以清晰看出,碳纳米管与导电炭黑一起在富锂锰基正极表面构筑的导电网络提升了电极的导电性、电极与电解液的浸润性,改善了电极/电解液界面反应。

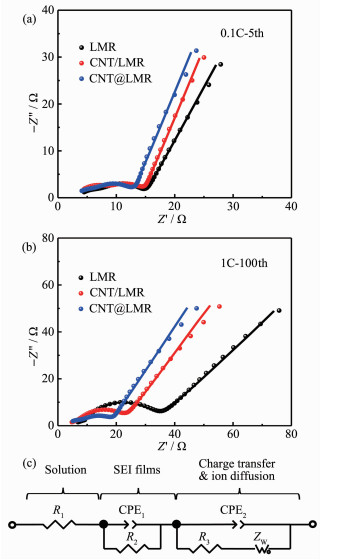
富锂锰基正极一般在小电流密度下充放电循环3次后,电极表面生成的SEI膜才能够稳定存在,且随着循环次数的不断增加,电极表面的SEI膜增厚和电极极化增大。因此,分别将LMR、CNT/LMR和CNT@LMR三种电极在0.1C倍率下充放电循环5次后和在1C倍率下充放电循环100次后进行了EIS测试。随后,构建等效电路模型对EIS测试谱图进行了拟合。图 6(a,b)分别是3种电极在循环5次和100次的EIS测试谱图(测试和拟合谱线),图 6(c)是拟合等效电路模型,表 1是EIS拟合参数。R1是溶液电阻,R2是电极SEI膜的离子扩散电阻,R3为电极/电解液界面的电荷转移电阻。CPE是具有电容效应的常相位角元件,即CPE的相位角不随频率变化而变化,只与指数P有关。用CPE代替电容C,是为了更好的拟合EIS谱线低频区的不规则半圆。CPE1-T和CPE2-T分别是EIS拟合前后2个半圆的容抗(电容对交流电的阻碍作用)。CPE1-P和CPE2-P表示拟合的修正指数P。当P值接近于1时,CPE的电容特性越强,即反应以电荷转移控制为主;当P值接近于0.5时,反应以离子扩散控制为主。对于韦伯扩散阻抗(ZW)来说,WR是韦伯扩散系数,ZW与WR成正比关系。WT是指扩散深度与颗粒扩散系数的比值,WP是韦伯扩散阻抗拟合的修正指数。
 下载:
导出CSV
下载:
导出CSV
| Sample | R1/W | SEI films | Charge transfer | Warburg diffusion | ||||||||
| R2/W | CPE1-T/mF | CPE1-P | R3/W | CPE2-T/mF | CPE2-P | WR | WT | WP | ||||
| LMR-5th | 3.19 | 6.59 | 0.12 | 0.62 | 5.75 | 0.1 | 0.87 | 2.71 | 0.004 | 0.37 | ||
| CNT/LMR-5th | 3.38 | 5.43 | 0.04 | 0.69 | 4.08 | 0.1 | 0.92 | 2.74 | 0.006 | 0.4 | ||
| CNT@LMR-5th | 3.18 | 4.21 | 0.12 | 0.63 | 5.11 | 0.11 | 0.85 | 2.08 | 0.004 | 0.4 | ||
| LMR-100th | 3.25 | 7.07 | 0.23 | 0.48 | 22.99 | 0.05 | 0.86 | 366.6 | 9.05 | 0.55 | ||
| CNT/LMR-100th | 3.36 | 5.5 | 0.67 | 0.48 | 14.37 | 0.09 | 0.85 | 314.3 | 3.92 | 0.66 | ||
| CNT@LMR-100th | 3.44 | 5.12 | 0.13 | 0.58 | 7.31 | 0.11 | 0.85 | 109.1 | 1.33 | 0.34 | ||
结合图 6(a)和表 1数据可知,3种电极的溶液电阻R1基本相同,这保证了3种电极的EIS曲线拟合数据的一致性和可比性。LMR、CNT/LMR和CNT@ LMR电极的R2分别是6.59、5.43和4.21 Ω,说明碳纳米管改善了富锂锰基正极材料表面的电极/电解液界面反应,有效地降低了SEI膜的厚度和电阻,有利于锂离子在电极表面的固相扩散。CNT@LMR电极的R3为5.11 Ω,低于LMR电极的5.75 Ω,表明了碳纳米管增大了电极表面区域并提升了富锂锰基正极的电子电导特性,进而提高了富锂锰基正极的倍率性能。通过图 6(b)和表 1数据来观察3种电极在循环100次后的电化学阻抗的变化,进而探究碳纳米管改性富锂锰基正极材料的循环寿命好的原因。与图 6(a)相比,膜阻抗、电荷转移阻抗和韦伯扩散阻抗均有较大增加,这是因为随着电极循环次数不断增加,电解液的消耗、电极表面副产物的增加和电极材料内部结构的变化等导致的电极极化增大。图 6(b)表明CNT@LMR的电化学阻抗谱线中第1个半圆和第2个半圆明显小于另外2个电极。在第1个半圆所代表的R2数值中,CNT@LMR电极的值为最小的5.12 Ω。在第2个半圆所代表的R3中,LMR、CNT/LMR和CNT@LMR电极的R3值分别是22.99、14.37和7.31 Ω,体现了碳纳米管改性对富锂锰基正极的极化降低和促进其电荷转移的作用。最后对比3种电极的韦伯扩散系数(WR),CNT@LMR电极的数值为最小,证明了在低频交流阻抗测试条件下,碳纳米管改性的富锂锰基正极材料拥有最优的离子扩散特性。
采用了一种新策略实现了碳纳米管对富锂锰基正极材料Li1.184[Ni0.15Mn0.516Co0.15]O2的表面和内部的复合改性。碳纳米管在材料表面和内部构筑的导电网络,有效地提升了材料的电子电导能力,大幅度提高了富锂锰基正极的倍率性能。在5C倍率放电下,碳纳米管改性的富锂锰基正极材料的比容量可达到141.4 mAh·g-1。碳纳米管在富锂锰基正极材料表面的致密网状结构改善了富锂锰基正极/电解液的界面反应,抑制了SEI膜的生长并减缓了电极极化,从而提升了富锂锰基正极材料的容量循环保持率。碳纳米管改性的富锂锰基正极材料在1C倍率下循环100次后,容量保持率为87.2%。
Chu S, Cui Y, Liu N. Nat. Mater., 2017, 16(1):16
Li M, Lu J, Chen Z W, et al. Adv. Mater., 2018, 30:1800561 doi: 10.1002/adma.201800561
Goodenough J B, Park K S. J. Am. Chem. Soc., 2013,135(4):1167-1176 doi: 10.1021/ja3091438
严武渭, 柳永宁, 崇少坤, 等.化学进展, 2017, 29(2/3):198-209YAN Wu-Wei, LIU Yong-Ning, CHONG Shao-Kun, et al. Progress in Chemistry, 2017, 29(2/3):198-209
Nayak P K, Erickson E M, Schipper F, et al. Adv. Energy Mater., 2018, 8(8):1702397 doi: 10.1002/aenm.201702397
Zheng J M, Myeong S, Cho W, et al. Adv. Energy Mater., 2016, 7:1601284
Li W D, Song B H, Manthiram A. Chem. Soc. Rev., 2017, 46(10):3006-3059 doi: 10.1039/C6CS00875E
Zheng J M, Xu P H, Gu M, et al. Chem. Mater., 2015, 27(4):1381-1390
Hu E Y, Yu X Q, Lin R Q, et al. Nat. Energy, 2018, 3(8):690-698 doi: 10.1038/s41560-018-0207-z
Pang S L, Wang Y G, Chen T, et al. Ceram. Int., 2016, 42(4):5397-5402 doi: 10.1016/j.ceramint.2015.12.076
Xiao B W, Sun X L. Adv. Energy Mater., 2018, 8(29):1802057 doi: 10.1002/aenm.201802057
Park K J, Lee B S, Park J H, et al. Phys. Chem. Chem. Phys., 2016, 18(23):15861-15866 doi: 10.1039/C6CP02007K
Chen D R, Zheng F, Li L, et al. J. Power Sources, 2017,341:147-155 doi: 10.1016/j.jpowsour.2016.11.020
Zheng F H, Deng Q, Zhong W T, et al. ACS Sustainable Chem. Eng., 2018, 6(12):16399-16411 doi: 10.1021/acssuschemeng.8b03442
Li H Q, Zhou H S. Chem. Commun., 2012, 48(9):1201-1217 doi: 10.1039/C1CC14764A
Lee S W, Yabuuchi N, Gallant B M, et al. Nat. Nanotech., 2010, 5(7):531 doi: 10.1038/nnano.2010.116
Liu X M, Huang Z D, Oh S W, et al. Compos. Sci. Technol., 2012, 72(2):121-144
Yuan W Y, Zhang Y, Cheng L F, et al. J. Mater. Chem. A, 2016, 4(23):8932-8951 doi: 10.1039/C6TA01546H
Guo L C, Zhao N Q, Li J J, et al. ACS Appl. Mater. Interfaces, 2015, 7(1):391-399 doi: 10.1021/am506354e
尹艳萍, 卢华权, 王忠, 等.无机化学学报, 2015, 31(10):1966-1970 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20151007&flag=1YIN Yan-Ping, LU Hua-Quan, WANG Zhong, et al. Chinese J. Inorg. Chem., 2015, 31(10):1966-1970 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20151007&flag=1
Wang Z, Yin Y P, Ren Y, et al. Nano Energy, 2017, 31:247-257 doi: 10.1016/j.nanoen.2016.10.014
Johnson C S, Li N C, Lefief C, et al. Electrochem. Commun., 2007, 9(4):787-795
Toprakci O, Toprakci H A, Li Y, et al. J. Power Sources, 2013,241:522-528 doi: 10.1016/j.jpowsour.2013.04.155
Weill F, Tran N, Croguennec L, et al. J. Power Sources, 2007,172(2):893-900 doi: 10.1016/j.jpowsour.2007.05.090
Gong Z L, Liu H S, Guo X J, et al. J. Power Sources, 2004,136(1):139-144 doi: 10.1016/j.jpowsour.2004.05.022
Ruther R E, Dixit H, Pezeshki A M, et al. J. Phys. Chem. C, 2015,119(32):18022-18029 doi: 10.1021/acs.jpcc.5b03900
陈丹丹, 李广社, 范建明, 等.无机化学学报, 2018, 34(4):703-711 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180413&flag=1CHEN Dan-Dan, LI Guang-She, FAN Jian-Ming, et al. Chinese J. Inorg. Chem., 2018, 34(4):703-711 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20180413&flag=1
张莹莹, 张锦.化学学报, 2012, 70(22):2293-2305ZHANG Ying-Ying, ZHANG Jin. Acta Chim. Sinica, 2012, 70(22):2293-2305
Lim J S, Moon J, Gim J, et al. J. Mater. Chem., 2012, 22(23):11772-11777 doi: 10.1039/c2jm30962a
Lu Z H, Dahn J R. J. Electrochem. Soc., 2002,149(7):A815-A822 doi: 10.1149/1.1480014
Li X C, Zheng L L, Zang Z, et al. J. Alloys Compd., 2018,744:41-50 doi: 10.1016/j.jallcom.2018.02.067
Ma D T, Zhang P X, Li Y L, et al. Sci. Rep., 2015, 5:11257 doi: 10.1038/srep11257

图 1 (a) CNT@LMR制备流程的SEM图和(b)示意图
Figure 1 (a) SEM images and (b) schematic illustration of synthesis process of CNT@LMR

图 2 (a) LMR和(b) CNT@LMR的SEM图; (c) LMR和CNT@LMR的XRD图和(d) Raman图
Figure 2 (a) SEM images of the (a) LMR and (b) CNT@LMR; (c) XRD patterns and (d) Raman spectrum of LMR and CNT@LMR

图 3 (a) CNT@LMR一次颗粒的SEM图和(b) TEM图
Figure 3 (a) SEM and (b) TEM images of primary particle of CNT@LMR

图 4 富锂锰基材料改性前后的电化学测试
Figure 4 Electrochemical tests of LMR before and after modification
(a) First charge-discharge curves; (b) First CV curves; (c) Rate performance; (d) Cyclic capability performance

图 5 (a, c) LMR和(b, d) CNT@LMR的SEM图和示意图
Figure 5 SEM image and schematic illustration of (a, c) LMR and (b, d) CNT@LMR

图 6 (a, b)正极材料在不同测试条件下的EIS; (c)等效电路图
Figure 6 (a, b) EIS of electrodes at different test conditions; (c) Equivalent circuit diagram
表 1 富锂锰基正极改性前后的EIS拟合参数
Table 1. Fitting EIS parameters of LMR electrodes before and after modification
| Sample | R1/W | SEI films | Charge transfer | Warburg diffusion | ||||||||
| R2/W | CPE1-T/mF | CPE1-P | R3/W | CPE2-T/mF | CPE2-P | WR | WT | WP | ||||
| LMR-5th | 3.19 | 6.59 | 0.12 | 0.62 | 5.75 | 0.1 | 0.87 | 2.71 | 0.004 | 0.37 | ||
| CNT/LMR-5th | 3.38 | 5.43 | 0.04 | 0.69 | 4.08 | 0.1 | 0.92 | 2.74 | 0.006 | 0.4 | ||
| CNT@LMR-5th | 3.18 | 4.21 | 0.12 | 0.63 | 5.11 | 0.11 | 0.85 | 2.08 | 0.004 | 0.4 | ||
| LMR-100th | 3.25 | 7.07 | 0.23 | 0.48 | 22.99 | 0.05 | 0.86 | 366.6 | 9.05 | 0.55 | ||
| CNT/LMR-100th | 3.36 | 5.5 | 0.67 | 0.48 | 14.37 | 0.09 | 0.85 | 314.3 | 3.92 | 0.66 | ||
| CNT@LMR-100th | 3.44 | 5.12 | 0.13 | 0.58 | 7.31 | 0.11 | 0.85 | 109.1 | 1.33 | 0.34 | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: