
图 1.
光催化剂的XRD图
Figure 1.
XRD patterns of samples

近年来,随着经济的快速发展,日益枯竭的资源和严重的环境污染已成为全球人类面临的共同问题。以太阳能为基础的光催化技术可以将太阳能直接转化成化学能,实现清洁能源的转化以及对环境污染物的净化[1-2]。传统半导体光催化剂如TiO2、ZnO等已被广泛应用于有机污染物降解[3]、重金属离子还原[4]和光解水制氢[5]等方面。但是,高光生电子-空穴对复合率以及低太阳能利用率等缺点限制了其实际应用价值[6-9]。因此,设计开发新型高效且具有宽可见光响应的光催化半导体材料成为科研工作者重要的研究方向。
石墨相碳氮化物(g-C3N4,主要组成元素C、N)近年来由于其合适的带隙、低廉的成本、优异的稳定性等,已成为非金属光催化剂的研究热点,可用于光解水析氢析氧、有机污染物分解、CO2还原和抗菌等[10-12]。然而,g-C3N4限于聚合物本质,具有较短的激子寿命,光催化性能不理想,因此仍需大量的改性措施来提高其光催化性能。目前,模板法合成(SiO2、SBA-15等)[13-14]、掺杂(B、S和Fe等)[15-16]、贵金属修饰(Au、Pt和Ag等)[17]及半导体复合(AgX、ZnO、CdS等)[18-19]等方法普遍用于g-C3N4的结构改性,在一定程度上均可提高其光催化活性。
原位非金属元素掺杂在保持其非金属结构的基础上,可以利用不同非金属原子的电子结构调控半导体电荷分布,优化电荷传输性能,可大幅提高g-C3N4的光催化活性。目前,诸多研究报道表明非金属元素(如F、I、S、O等)掺杂能优化g-C3N4的能带结构,有效促进电荷重复,有利于光生载流子的分离[20]。氧(O)掺杂可实现在不引入其他元素的情况下,增加g-C3N4结构缺陷,在导带底部引入新的缺陷态能级,同时改变聚合结构的电荷重组,有利于调整g-C3N4的能带结构和光电性质,拓展可见光响应范围,有效提高对可见光的利用率。例如,Li等[21]采用简单的H2O2水热法将氧原子引入g-C3N4,成功合成了具有分级结构的O掺杂g-C3N4,有效扩大了表面积,增强了可见光响应,在制H2和甲基蓝(MB)的降解方面表现出优异的光活性。Huang等[22]将三聚氰胺和H2O2混合预处理后,通过热聚合法制造出具有新型多孔网络的O掺杂g-C3N4,显示出比块状g-C3N4高6.1倍的光解水制氢活性。
本研究以二聚氰胺(DCDA)、尿素为原料,草酸为氧掺杂源,按照不同质量比混合后经两步热聚合,制备出不同氧掺杂量的CNO纳米片。采用多种表征技术对催化剂结构进行分析,通过光解水制氢来评价催化剂的活性。结果表明,所制备的CNO纳米片具有显著增强的光解水制氢活性。
马弗炉(济南精密科学仪器仪表有限公司);磁力搅拌器(德国,IKA公司);300 W氙灯、光催化活性评价系统(北京中教金源科技有限公司)、光催化反应系统(北京中教金源科技有限公司)等。
DCDA、尿素、草酸、三乙醇胺等均为分析纯,购自国药集团化学试剂有限公司。
称取DCDA和尿素各4 g,与一定质量比的草酸混合溶解于20 mL超纯水中,完全溶解后将油浴升温至80 ℃,待固体干燥后研磨成粉末;将上述所得固体粉末置于加盖坩埚中,在马弗炉中550 ℃煅烧2 h(升温速率为5 ℃·min-1),冷却研磨,产物标记为CNO1-x。将产物进行二次煅烧,条件同上,冷却后研磨,样品标记为CNO2-x,x表示草酸与DCDA和尿素混合物的质量百分比,分别为10%、20%、30%、40%、50%。
在上述过程中不加入草酸,直接煅烧DCDA和尿素的混合物,其他步骤同上,所得产物为纯氮化碳,标记为CN1和CN2。没有特殊说明,文中CN和CNO样品均指CN2和CNO2。
采用XRD(D/max 2500PC,日本理学株式会社)分析材料成分,工作电压及电流分别为40 kV和30 mA,靶源为Cu Kα射线(波长为0.150 4 nm),扫描范围为5°~55°;利用SEM(Nova Nano SEM 230,美国FEI公司)、TEM(JEM-2100,日本理学株式会社)观察样品形貌,加速电压分别为5和200 kV;采用BET(JW-BK122W,精微高博)进行N2吸附-脱附测试,分析样品的比表面积及孔径分布;采用UV-Vis(UV-2550,日本Shimadzu公司)表征样品的光吸收性质,BaSO4为标准物质,测量范围200~800 nm;XPS(ESCALAB 250,美国Thermo Fisher Scientific公司)分析材料中元素的化学性质;PL(Quanta MasterTM40,美国PTI公司)考察催化剂的发光性能;利用电化学工作站(CHI660E,上海辰华仪器有限公司)表征催化剂的电化学性质。
50 mg催化剂分散于100 mL含有10%(V/V)的三乙醇胺的水溶液中,将100 μL氯铂酸水溶液(H2PtCl6·6H2O,0.04 g·mL-1)加入到上述溶液中, 并搅拌30 min。系统抽真空后使用300 W氙灯光源(400 nm cut滤光片)进行光照,使3%(w/w)的Pt原位光沉积在催化剂表面。每隔20 min通过气相色谱(TCD检测器,N2为载气)在线自动取样分析H2的产生量。
图 1为所制备催化剂的XRD图。CN具有2个典型的XRD衍射峰,分别对应于2θ=12.9°(d=0.68 nm)和27.6°(d=0.321 nm)处的石墨相氮化碳(100)和(002)晶面[23]。掺杂会引起物相晶格常数变化,使得晶面衍射峰的位置发生偏移。从图中可以看出,O掺杂后,CNO样品的(002)面衍射峰位向低角度明显偏移,说明O元素掺杂导致石墨层间距变大(0.328 nm)。同时,(002)面衍射峰强度明显降低,表明O掺杂降低了催化剂层间堆叠结构的长程有序度,这可能归因于O元素的掺杂引入结构缺陷,导致材料聚合度降低[24]。而(100)面衍射峰未发生明显变化,说明O掺杂未改变面内杂环的基本聚合结构。
图 2为所制备催化剂的SEM和TEM图。由图 2(a)可见热聚合法制备的CN为典型的块状分布,由紧密片层堆积而成。O掺杂后得到CNO的致密性明显降低,组成该催化剂的纳米片颗粒尺寸缩小(图 2(b)),这与O元素掺杂导致CN层间距扩大的XRD结果一致。图 2(c,d)分别是CN和CNO-40%的TEM图,掺杂后的CNO保持了石墨相的片状堆积。同时结合图 2(d)发现,样品由“丝绒”状纳米片无规则组合而成,同时多孔性明显增加。这是由于草酸在热缩聚过程中发生热分解,产生易挥发性气体作为“造孔剂”,降低了催化剂的聚合密度。同时,O元素的掺杂,在一定程度上降低了g-C3N4结构的堆积有序度。图 2(e,f)是CN和CNO的N2吸附-脱附测试结果,相比于CN的比表面积23 m2·g-1和孔体积0.115 m3·g-1,CNO分别提高至58 m2·g-1和0.202 m3·g-1,同样证明O掺杂导致g-C3N4聚合度降低,多孔性增加。多孔结构在比表面积增大的同时可为催化反应提供更多的活性位点,有利于催化反应速率的提高。
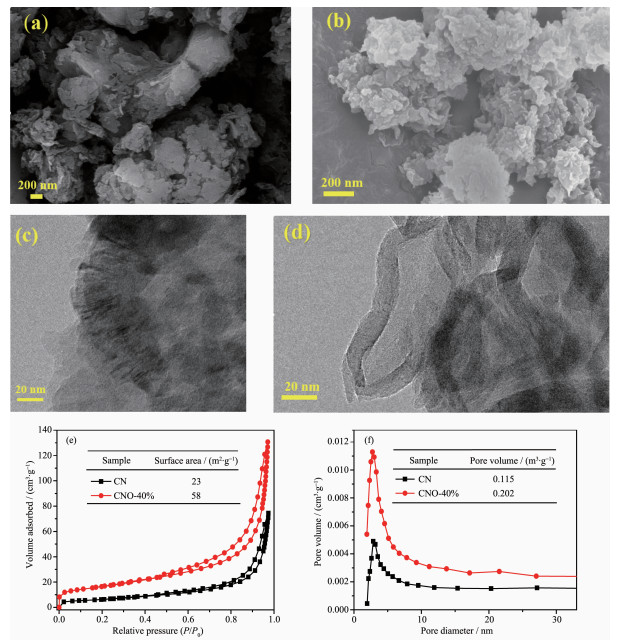
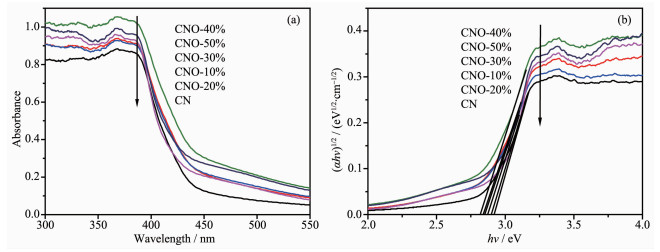
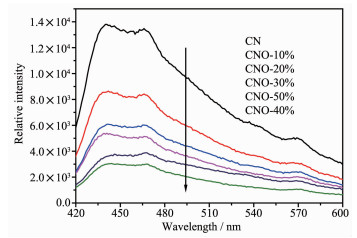
图 3是催化剂的UV-Vis光吸收谱图。由图 3(a)可见,CN的光吸收带边在450 nm附近[25]。经过O掺杂改性后的CNO光吸收带边发生明显红移,扩展至近480 nm,提高了对可见光的利用率。同时,相比于CN,一系列CNO样品随着掺杂量不断增加,对光的吸收性也不断提高,尤其是在可见光区(600 nm>λ>450 nm)。其中,CNO-40%表现出最强的光吸收性。根据Kubelka-Munk方程计算转化可得到样品的光学禁带宽度[26],结果如图 3(b)所示,CN的带隙值为2.92 eV,氧掺杂后样品的带隙值明显向低位移动,10%~50%样品的带隙值分别为2.88、2.90、2.85、2.82、2.84 eV。在宽可见光照射下即可产生光生载流子的跃迁,从而有利于提高光催化活性。
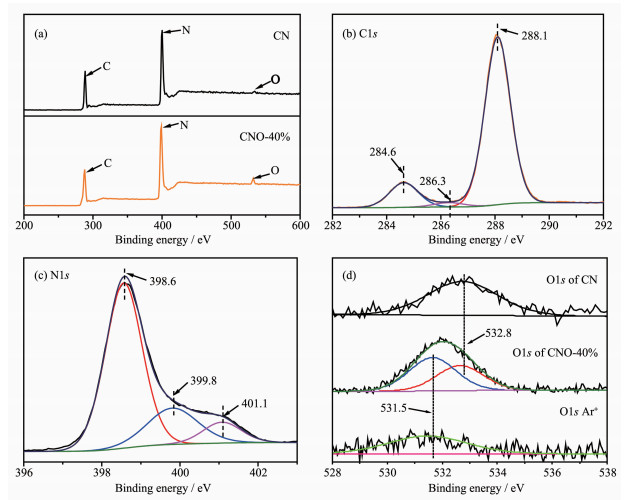
为了研究催化剂表面元素的化学状态,对样品CNO-40%进行了XPS分析。图 4(a)为CN和CNO-40%的XPS总谱图。结合表 1可知,掺杂前后C元素的含量几乎未发生改变,而N元素的含量由55.96%减少至53.87%,同时O元素含量由1.85%增加至4.11%,说明N缺陷的增加源于O原子取代了CN结构中部分N原子而掺杂至骨架结构中。
 下载:
导出CSV
下载:
导出CSV
| Sample | C/%(n/n) | N/%(n/n) | O/%(n/n) |
| CN | 42.19 | 55.96 | 1.85 |
| CNO-40% | 42.02 | 53.87 | 4.11 |
为进一步分析元素的键合状态,对C、N、O元素谱图进行进一步分析。如图 4(b)所示,C1s谱可拟合为284.6、286.3和288.1 eV三个特征峰,其中结合能284.6 eV处的峰归属于催化剂表面无定型C;286.3 eV峰可归因于C-O键的存在,表明杂化单元中的芳环O原子掺杂;结合能288.1 eV处的峰归属于CN三嗪环结构中的sp2杂化C原子(N-C=N)[27]。图 4(c)为N1s峰的拟合结果,主要存在398.6、399.8和401.1 eV三个峰,分别对应为sp2杂化的N原子(C-N=C)、与杂环结构相连的桥N原子[N-(C)3]和末端氨基中的N原子(C-N-H) [28]。C、N峰的分析结果表明,CNO样品基本保持C-N杂环结构,O元素的掺杂未显著改变其结构。O1s的XPS图谱如图 4(d)所示,CN中只含有1个结合能为532.8 eV的特征峰,对应于催化剂表面吸附水或氧。而CNO样品除了表面O峰,还增加了531.5 eV处的峰,意味着N-C-O/C-O键的产生[29],其含量占催化剂中O元素总含量的50.23%(n/n),为O掺杂g-C3N4催化剂的形成提供了直接证据。对CNO-40%样品表面进行Ar+刻蚀30 s去除表面层后,O1s中532.8 eV的峰完全消失,但531.5 eV处的峰仍然存在,证明掺杂O原子为晶格取代氧,均匀掺杂在骨架中,而不仅是表面O。综上结果表明O原子通过取代sp2杂化的N原子直接键合到与之相连的碳上,在C-N杂环骨架结构中形成O掺杂。
对催化剂的光解水制氢性能进行考察。由催化剂的产氢速率图 5(a)可知,光照时间1 h后,CN1的产氢速率13 μmol·h-1,经过二次煅烧后提高至22.6 μmol·h-1,可见二次煅烧热剥离是提高氮化碳光催化活性的有效途径。O掺杂后的所有CNO1样品的产氢速率与CN1相比进一步得到提高,CNO1-40%的产氢速率达28.7 μmol·h-1,因此氧掺杂有利于氮化碳制氢活性的提高。在此基础上,将O掺杂催化剂进行二次煅烧热处理,所得CNO2产物的产氢活性均显著得到提高,且随着O掺杂含量的增加,制氢活性逐渐提高,其中CNO2-40%的产氢速率达88.6 μmol·h-1,是CN2的3.9倍,CN1的6.8倍。在单色光照下(420 nm)测定CNO-40%产氢的量子效率为0.96%。而CNO-50%的制氢活性反而降低,可能是由于过量O掺杂引起聚合结构无序度的增加,降低了电荷的有效传输性。同时,所有催化剂在连续光照条件下,其产氢量随着光照时间基本呈线性增加,说明了其良好的光催化响应活性(图 5(b))。
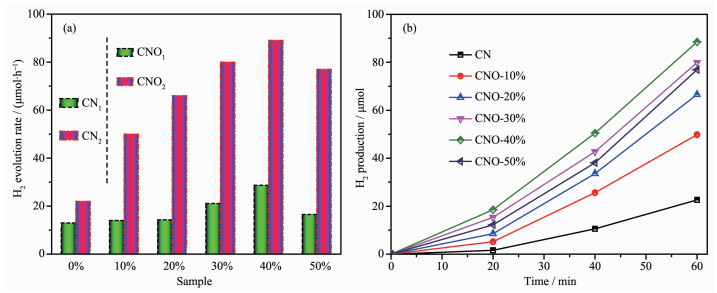
(a) H2 evolution rate; (b) Cumulative amount of H2 with light irradiation time
图 6(a)为CNO-40%光催化水解制氢多次循环使用测试的结果。在第2次使用时产氢量有轻微降低,经过连续多次循环使用后,H2产量虽有轻微减少但基本保持稳定,说明该催化剂具有较好的产氢稳定性,且产氢量始终随光照时间呈线性增长。
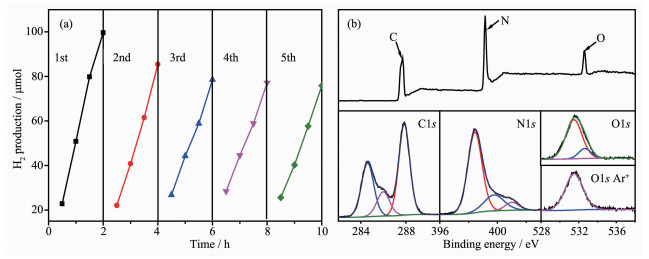
图 6(b)为对反应后的CNO-40%进行的XPS测试结果。由图可知,反应后O元素含量明显增加,由反应前4.11%增至8.86%。同时对比各元素反应前后的图谱发现,N1s图谱几乎未发生变化,C1s图谱中C-O键对应的峰值明显增强,同时元素刻蚀证明O主要归于骨架掺杂形成的N-C-O/C-O键,反应后并未发生明显降低,说明催化剂主体结构未发生变化。反应初期产氢速率的略微下降可能是由于催化剂表面水化,活性位点数降低。当液固界面达到平衡态,活性稳定性基本保持。
光生电荷的分离/复合效率是影响光催化剂的性能的重要因素之一,电子-空穴对的高效分离有利于光催化反应有效进行。图 7是催化剂的稳态PL发射谱图,可见未掺杂的CN在440~470 nm有较强的PL发射峰,来源于催化剂受光激发后产生的带边荧光现象。经O掺杂后,样品PL发射图谱的位置未发生变化,但发射峰强度随着O掺杂量的增加发生明显降低,说明O元素掺杂后在很大程度上降低了光生电子-空穴对的复合率。其中,CNO-40%的PL猝灭现象最明显。Huang等[22]利用密度泛函理论(DFT)计算证明,O掺杂最易取代CN结构中二配位的N位点,而周围多余的电子会被重新分配到相邻的C原子中,并在大π键中进行离域,从而导致g-C3N4的导带下方产生一个新的缺陷态,进而提高g-C3N4对可见光的利用率,同时促进电子-空穴对的分离。
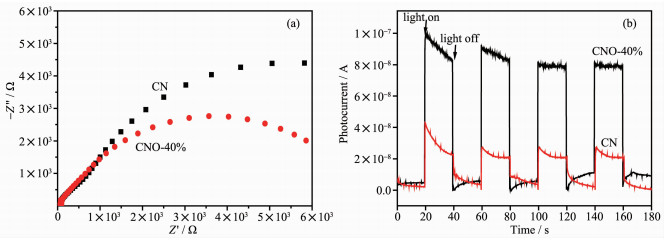
电化学阻抗往往用来直观表示电荷在半导体中的传输性能,其阻抗谱图(EIS)半径越大说明电荷传输电阻越大,载流子复合率越高。图 8(a)为CN和CNO-40%样品的EIS谱图,由图可见相比于纯CN,O掺杂后的CNO圆弧半径明显减小,说明O元素的掺杂有利于降低传输电阻,证实O掺杂可提高电荷的迁移速率,降低电子-空穴对的复合率。光电流测试同样被用来评估光生电子-空穴对的分离效率,结果如图 8(b)所示。在瞬间光照条件下,CN和CNO均能够快速响应产生光电流,说明其较好的光电转化性能。其中,CNO光电流响应值远远高于CN,说明在光照条件下,O掺杂改性有利于光生电荷的产生,同时结合PL测试及阻抗分析,O掺杂后的杂化结构有利于加速光生电荷的迁移,使得光生电子-空穴对有效分离,从而提高催化剂的活性。
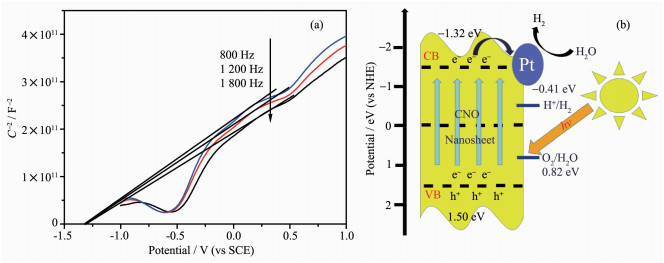
根据莫特-肖特曲线确定CNO-40%的导带(CB)电位为-1.32 eV (图 9(a)),远高于H2O的还原电位-0.41 eV(pH=7)。结合带隙大小确定CNO-40%价带(VB)电位为1.50 eV,H2O的氧化电位0.82 eV。由图 9(b)可知,催化剂受到足够能量的光激发,光生电子从价带跃迁到导带,在Pt助催化剂的辅助下参与还原反应分解水释放H2。
采用二聚氰胺和尿素为原料,草酸为氧源,通过热聚合法二步合成氧掺杂氮化碳纳米片催化剂,并考察了其光催化分解水制氢性能。结果表明,二次热处理能够提高催化剂的多孔性,同时保持O原子掺杂。掺杂的O原子取代C-N杂环中的N原子直接键合到三嗪环结构中sp2杂化的碳上。掺杂后的CNO催化剂吸收边带红移至480 nm,光吸收能力增强,同时电子-空穴对复合率显著降低。在可见光照射下,二次热处理后的催化剂CNO-40%的分解水制氢活性最高,达88.6 μmol·h-1,是未掺杂CN的3.9倍,且具有优良的稳定性。CNO-40%产氢的量子效率为0.96%(420 nm)。这项工作表明经过简单地预处理前驱体不仅可以调控结构,还可以引入有用的杂原子甚至单体。
Cates E L, Chinnapongse S L, Kim J H, et al. Environ. Sci. Technol., 2012, 46(22):12316-12328 doi: 10.1021/es303612p
Marin M L, Santos-Juanes L, Arques A, et al. Chem. Rev., 2012, 112(3):1710-1760 doi: 10.1021/cr2000543
Wang Y, Feng C X, Zhang M, et al. Appl. Catal. B, 2011, 104(3):268-274 https://www.sciencedirect.com/science/article/pii/S0926337311001330
Zhao D, Chen C S, Yu C L, et al. J. Phys. Chem. C, 2009, 113(30):13160-13165 doi: 10.1021/jp9002774
Zhao L, Lin X Z, Lai H B, et al. J. Mol. Catal., 2014, 28(3):276-281 https://www.researchgate.net/publication/289574300_Preparation_and_visible_light_photocatalytic_activity_of_silk_fibroinTiO2_nanocomposite?_sg=g2g7rLvAHPYLxyhCW60TYrGWd8zbFzEL3BbEIHuGoD2SJnVPTn64kuJoJQ-qK1g9vDGaTWZmOzmF8tQ3cVaKbQ
黄涛, 张国亮, 张慧, 等.化工进展, 2010, 29(3):498-504 http://www.cnki.com.cn/Article/CJFDTotal-HGJZ201003024.htmHUANG Tao, ZHANG Guo-Liang, ZHANG Hui, et al. Chemical Industry and Engineering Progress, 2010, 29(3):498-504 http://www.cnki.com.cn/Article/CJFDTotal-HGJZ201003024.htm
张晶, 张亚萍, 于濂清, 等.无机盐工业, 2010, 42(9):6-9 doi: 10.3969/j.issn.1006-4990.2010.09.003ZHANG Jing, ZHANG Ya-Ping, YU Lian-Qing, et al. Inorganic Chemicals Industry, 2010, 42(9):6-9 doi: 10.3969/j.issn.1006-4990.2010.09.003
倪广红, 丰平.纳米科技, 2010, 7(2):81-86NI Guang-Hong, FENG Ping. Nanoscience & Technology, 2010, 7(2):81-86
张亚萍, 张安玉, 于濂清, 等.无机材料学报, 2016, 31(3):269-272 http://www.cnki.com.cn/article/cjfdtotal-wgcl201603007.htmZHANG Ya-Ping, ZHANG An-Yu, YU Lian-Qing, et al. J. Inorg. Mater., 2016, 31(3):269-272 http://www.cnki.com.cn/article/cjfdtotal-wgcl201603007.htm
Cao S, Low J, Yu J, et al. Adv. Mater., 2015, 27(13):2150-2176 doi: 10.1002/adma.201500033
Liu J H, Xie S Y, Geng Z B, et al. Nano Lett., 2016, 16(10):6568-6573 doi: 10.1021/acs.nanolett.6b03229
陈艳, 刘海波, 等.无机化学学报, 2017, 33(12):2255-2261 doi: 10.11862/CJIC.2017.218CHEN Yan, LIU Hai-Bo, et al. Chinese J. Inorg. Chem., 2017, 33(12):2255-2261 doi: 10.11862/CJIC.2017.218
Li X H, Wang X C, Antonietti M, et al. J. Am. Chem. Soc., 2009, 131(5):1680-1681 doi: 10.1021/ja809307s
Li X H, Wang X C, Antonietti M, et al. Chem. Sci., 2012, 3(6):2170-2174 doi: 10.1039/c2sc20289a
Yan S C, Li Z S, Zou Z G, et al. Langmuir, 2010, 26(6):3894-3901 doi: 10.1021/la904023j
Liu G, Niu P, Sun C H, et al. J. Am. Chem. Soc., 2010, 132(33):11642-11648 doi: 10.1021/ja103798k
Yan D D, Dr X W, Thomas P, et al. ChemCatChem, 2010, 2(7):834-838 doi: 10.1002/cctc.201000057
Liu Y A, Wang R X, Yang Z K, et al. Chin. J. Catal., 2015, 36(12):2135-2144 doi: 10.1016/S1872-2067(15)60985-8
Jie F, Chang B B, Tian Y L, et al. J. Mater. Chem. A, 2013, 1(9):3083-3090 doi: 10.1039/c2ta00672c
Ong W J, Tan L L, Ng Y H, et al. Chem. Rev., 2016, 116(12):7159-7329 doi: 10.1021/acs.chemrev.6b00075
Li J H, Shen B, Hong Z H, et al. Chem. Commun., 2012, 48(98):12017-12019 doi: 10.1039/c2cc35862j
Huang Z F, Song J, Pan L, et al. Nano Energy, 2015, 12:646-656 doi: 10.1016/j.nanoen.2015.01.043
崔言娟, 王愉雄, 王浩, 等.催化学报, 2016, 37(11):1899-1906 http://www.cnki.com.cn/Article/CJFDTotal-CHUA201611012.htmCUI Yan-Juan, WANG Yu-Xiong, WANG Hao, et al. Chin. J. Catal., 2016, 37(11):1899-1906 http://www.cnki.com.cn/Article/CJFDTotal-CHUA201611012.htm
Rong X S, Qiu F X, Rong J, et al. J. Solid State Chem., 2015, 230:126-134 doi: 10.1016/j.jssc.2015.07.003
翟顺成, 郭平, 郑继明, 等.物理学报, 2017, 66(18):187102 doi: 10.7498/aps.66.187102ZHAI Shun-Chen, GUO Pin, ZHEN Ji-Ming, et al. Acta Physica Sinica, 2017, 66(18):187102 doi: 10.7498/aps.66.187102
李欣, 王铁成, 屈广州, 等.环境工程学报, 2017, 11(5):2738-2742 http://www.cnki.com.cn/Article/CJFDTotal-HJJZ201705017.htmLI Xin, WANG Tie-Cheng, QU Guang-Zhou, et al. Chinese Journal of Environmental Engineering, 2017, 11(5):2738-2742 http://www.cnki.com.cn/Article/CJFDTotal-HJJZ201705017.htm
Fu Y S, Zhu J W, Hu C, et al. Nanoscale, 2014, 6(21):12555-12564 doi: 10.1039/C4NR03145H
Oh J, Yoo R J, Kim S Y, et al. Chem. Eur. J., 2015, 21(16):6241-6246 doi: 10.1002/chem.v21.16
Li J H, Shen B, Hong Z H, et al. Chem. Commun., 2012, 48(98):12017-12019 doi: 10.1039/c2cc35862j

图 2 CN和CNO-40%的SEM图(a, b), CN和CNO-40%的TEM图(c, d), N2吸附-脱附等温线(e)和BJH孔径分布(f)
Figure 2 SEM images of CN and CNO-40% (a, b), TEM images of CN and CNO-40% (c, d), N2 adsorption-desorption isotherms (e) and the corresponding BJH pore-size distribution (f)

图 3 样品的(a)紫外-可见漫反射吸收光谱和(b)禁带宽度值
Figure 3 (a) UV-Vis diffuse reflectance absorbance spectra and (b) band gap values of samples

图 4 催化剂CN和CNO-40%的XPS总谱(a); CNO-40%中C1s和N1s谱图(b, c); CN和CNO-40%及其刻蚀30 s后O1s的XPS谱图(d)
Figure 4 XPS total spectra of CN and CNO-40% (a); C1s and N1s spectra of CNO-40% (b, c), O1s spectra of CN, CNO-40% and CNO-40% after 30 s etching (d)

图 5 催化剂的光解水制氢性能
Figure 5 Photocatalytic H2 production of catalysts
(a) H2 evolution rate; (b) Cumulative amount of H2 with light irradiation time

图 6 CNO-40%的连续产H2稳定性(a)和反应后样品的XPS图谱(b)
Figure 6 H2 production stability of CNO-40% (a) and XPS spectra of the sample after the reaction (b)

图 8 CN和CNO-40%的阻抗图谱(a)和光电流图谱(b)
Figure 8 EIS (a) and photocurrent spectra (b) of CN and CNO-40%

图 9 CNO-40%的莫特-肖特曲线(a)和产氢机理图(b)
Figure 9 Mott-Schottky plots of CNO-40% (a) and mechanism diagram of hydrogen production (b)
表 1 样品中C、N、O元素的含量
Table 1. Content of C, N and O in the samples
| Sample | C/%(n/n) | N/%(n/n) | O/%(n/n) |
| CN | 42.19 | 55.96 | 1.85 |
| CNO-40% | 42.02 | 53.87 | 4.11 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: