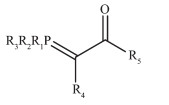
Figure 1.
Structure of phosphorous ylide
X-ray Crystallography, Spectral Characterization and Computational Studies of Mercury(Ⅱ) Complex with 1-(p-Tolyl)-2-(tri-p-tolyl-λ5-phosphanylidene)ethan-1-one
Pourmirza Mahsa , Ebrahimnezhad Shahla , Ramazani Ali , Bahrami Aidin , Alaei Sholeh. , Reza Dadrass Ali.

Ylides play an important role in the production of the chemical compounds, because ylides are a fundamental part of the structure of compounds which contain biological, pharmaceutical and industrial properties[1-3]. In ylides, carbanion directly attaches to one heteroatom with positive charge. Phosphorous ylides are formed when phosphorus enters to the structure of ylide. Because of resonance forms produc-tion, presence of carbonyl groups in adjacency of methylene carbon will lead to the higher stability of ylides. In addition to the biological, pharmaceutical and industrial applications[4-6], phosphorous ylides can coordinate with metal ether by the oxygen of carbonyl group or by methine carbon.
These compounds as ambidentate ligands hold substantial place in the organometallic chemistry. Each year considerable numbers of papers are published in this regard[7-9]. Although these complexes have some catalytic, biologic and pharmaceutical properties, their main importance is about their theoretical aspects, especially the ability of having comprehensive study on the competition of connection of hard oxygen or soft carbon in order to coordination to the hard, soft metal and transition metal centers. The comprehensive study of these complexes is so important, because there is flexibility in the structure of these compounds where R1 to R5 can be changed selectively[10].
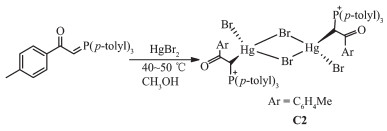
In this paper, the preparation of mercury(Ⅱ) and cadmium(Ⅱ) complexes with 1-(p-tolyl)-2-(tri-p-tolyl-λ5 -phosphanylidene)ethan-1-one(L)[11] will be discussed. The crystals of [Hg(L)(μ2-Br)Br]2 (C2) were obtained from the reaction between ligand L and HgBr2, and the crystal was characterized by X-ray crystallography and IR, 1H NMR, 31P NMR, 13C NMR spectroscopic methods. Since mercury(Ⅱ) and cadmium(Ⅱ) are soft Lewis acid, their attachment to phosphorous ylide is from soft side which is methine carbon and can be confirmed by analytical methods. Since mercury is a transition metal and has full d orbitals with large atomic radius, it is highly reactive with ylides especially phosphorous ylides to form organometallic complexes[12-13]. Theoretical calculations will determine the active site of complex C2 to bond with Schiff base. There are numerous reports about biological activities in ligands and metal complexes[9-10, 13]. Therefore, the titled compounds also can be a good candidate for biological research.
Petroleum benzene was distilled over sodium pieces prior to use. Ligand L was synthesized accor-ding to the reported method[11]. All other solvents were purchased from Merck Company and used without further purification. The 1H, 31P, 13C NMR spectra were recorded at 400.13, 161.98, and 100.62 MHz, respe-ctively on BRUKER spectrometer (Germany) in CDCl3 solvent. Solid state IR spectra in the region of 500~4 000 cm-1 using KBr pellets were obtained on a FT-IR Nexus 670 spectrophotometer. Melting points were determined on a digital melting point apparatus (Electrothermal 9100) and remained uncorrected.
Methanol solution (10 mL) of CdCl2 (924 mg, 0.5 mmol) was added to a solution of ligand L (218 mg, 0.5 mmol) in the same solvent, and the mixture was stirred for 18 h. The pale-yellow precipitate was filtered off, washed with petroleum benzene (2×20 mL), and dried. Yield: 83%. m.p. 182~184 ℃. FT-IR (KBr, cm-1): 1 631 (CO), 815 (P+-C). 1H NMR (DMSO-d6): δ 2.14 (s, 24H, CH3), 4.78 (d, 2JPH=10.44 Hz, 2H, CH), 7.49~8.26 (m, 32H, 8Ph). 13C NMR (DMSO-d6): δ 20.25 (CH3), 69.28 (CH), 128.75 (d, 1JP-C=128.87 Hz, PPh3(i)), 129.98 (COPh)(m)), 130.22 (d, 3JP-C=15.29 Hz, PPh3)(m)), 130.91 (COPh)(o)), 132.20 (d, 2JP-C=18.11 Hz, PPh3)(o)), 132.85(d, 3JP-C=15.29 Hz, COPh)(i)), 134.95 (s, PPh3)(p)), 143.60 (COPh)(p)), 196.88 (s, CO). 31P NMR (DMSO-d6): δ 30.38.
Methanol solution (10 mL) of HgBr2 (181 mg, 0.5 mmol) was added to a solution of ligand L (218 mg, 0.5 mmol) in methanol (15 mL) and the mixture was stirred for 24 h. The white precipitate was filtered off, washed with petroleum benzene (2×15 mL), and dried. To get the crystals of complex C2, ligand L 0.25 mmol (109 mg) in methanol (6 mL) were added dropwise to a solution of HgBr2 (0.25 mmol, 90 mg) in the same solvent (2 mL). After standing for several days without stirring at room temperature, colorless crystals were obtained. Yield: 80%. m.p. 206~208 ℃. FT-IR (KBr, cm-1): 1 626 (CO), 811 (P+-C). 1H NMR (CDCl3): δ 2.41 (s, 24H, CH3), 5.30 (d, J=12.41 Hz, 2H, CH), 7.25~8.02 (m, 32H, 8Ph). 13C NMR (CDCl3): δ 21.63 (CH3), 67.09 (CH), 127.55 (d, 1JP-C=124.87 Hz, PPh3)(i)), 128.70 (COPh)(m)), 129.25 (d, 3JP-C=12.58 Hz, PPh3)(m)), 129.76 (COPh)(o)), 131.25 (d, 2JP-C=13.68 Hz, PPh3)(o)), 132.18(d, 3JP-C=10.36 Hz, COPh)(i)), 133.74 (s, PPh3)(p)), 142.42 (COPh)(p)), 193.26 (CO). 31P NMR (CDCl3): δ 30.72.
The crystallographic measurement of C2 was performed on a Xcalibur R κ-geometry automated four-circle diffractometer equipped with a ruby CCD camera and graphite-monochromatized Mo Kα radiation (λ=0.071 073 nm). The data were collected at 90(2) K by using the Oxford-Cryosystems cooler. Data were corrected for Lorentz and polarization effects. Data collection, cell refinement, data reduction, and analysis were carried out with Xcalibur PX software, CrysAlisPro[14]. Because C2 was isomorphous with Hg2I4((p-tolyl)3PCHC(O)-C6H4Cl)2[15], the refinement of its structure was started by using the coordinates of non-H atoms taken from iodide derivative (Cambridge Structural Database (CSD, Version 5.35[16]). The stru-cture was refined by a full-matrix least-squares technique with SHELXL-2013[17] and anisotropic thermal parameters for non-H atoms. All H atoms were included from geometry and were refined using a riding model, with C-H bond length of 0.095~0.100 nm, and with Uiso(H)=1.2Ueq(C) for CH, 1.5Ueq(C) for CH3, respectively. The figures were drawn with the Diamond program[18]. Details of the conditions for the data collection and the structures refinements are given in Table 1.
 下载:
导出CSV
下载:
导出CSV
| Chemical formula | C60H58Br4Hg2O2P2 | Z | 2 | |
| Mr | 1 593.82 | Crystal size / mm | 0.30×0.27×0.21 | |
| Crystal system | Monoclinic | μ / mm-1 | 8.31 | |
| Space group | P21/c | Measured, independent, observed [I>2σ(I)] reflection | 21 419, 8 434, 7 453 | |
| a / nm | 1.178 9(2) | Rint | 0.022 | |
| b / nm | 1.078 3(2) | R1, wR2 | 0.023, 0.046 | |
| c / nm | 2.244 7(4) | S | 1.10 | |
| β / (°) | 95.63(2) | Parameter | 320 | |
| V / nm3 | 2.839 7(9) | Restraint | 0 | |
| Dc / (g·cm-3) | 1.864 | (Δρ)max, (Δρ)min / (e·nm-3) | 690, -650 |
CCDC: 1437693, C2.
Density functional theory (DFT)[19] calculations were accomplished using the GAMESS program package[20]. In the case of complex C2, the X-ray structure was considered as starting points for the geometrical investigations. The calculations were based on the B3LYP/6-31G* level of theory[21] for all atoms to provide the most stable structure.
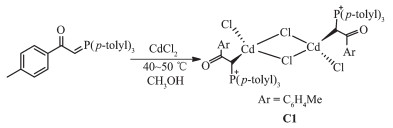
The reaction of tri-p-tolylphosphine with 2-bromo -1-(p-tolyl)ethanone at room temperature for 4 h (1:1 molar ratio) in acetone gave white solid of phosphonium salt in high yield. Further treatment with aqueous NaOH solution led to elimination of HBr, giving the free ligand L[11] (Scheme 1). Hg (Ⅱ) and Cd (Ⅱ) ions reacted with L (1:1 molar ratio) to produce complexes C1 and C2 (Scheme 2 and 3). By reviewing complex C1 spectroscopic data, investigating other similar compounds of Cd(Ⅱ)[22-23] and also considering that Cd and Hg share similar properties, the structure of complex C1 might be similar to C2 or other mercury halide-bridged dimeric complexes with phosphorous ylides[15, 24-25].
The infrared spectra of complexes C1 and C2 in the solid-state showed νCO in the range of 1 631 and 1 626 cm-1, indicating coordination of L through carbon at higher wave numbers with respect to the free L in 1 599 cm-1 [11]. The expected spectra of 31P and CH proton of C1 and C2 were shifted downfield as a consequence of C-coordination character of free L (CH in 4.36, 31P in 12.98)[11]. The 13C NMR shifts of CO group in C1 and C2 were higher than 191.34 noted for the same carbon in free L, indicating lower shielding of CO group carbon in the complexes.
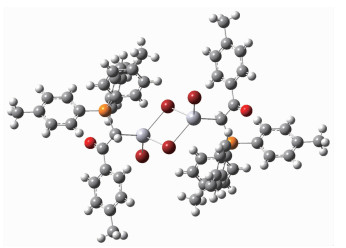
The X-ray crystallography analysis for crystal of complex C2 revealed that C2 crystalize in monoclinic systems. The molecular structure is shown in Fig. 2, relevant bond distances and angles are given in Table 2, and also geometry of hydrogen bonds in complex C2 is presented in Table 3. The X-ray analysis reveals that Br1 is more strongly bounded to Hg atom (Hg-Br1 0.254 77(5) nm) than Br2 (Hg-Br2 0.267 49(5) nm). This state demonstrates terminal bond is stronger than bridge bond. Compared to Hg-Br distances found in similar Hg(Ⅱ)-Br complexes[26-28], Hg-Br bonds in C2 are relatively stronger. The Hg(Ⅱ) in C2 is sp3 hybrid-ization and has a tetrahedral coordination. The C1-Hg1-Br bond angles (116.68(6)°, 126.06(6)°, 98.91(6)) and Br-Hg1-Br bond angles (108.250(16)°, 117.084(12)°, 85.139(17)°) indicate a relatively symmetric tetragonal environment around Hg(Ⅱ) ion. The comparison of bond angles between Br2-Hg1-Br2ⅰ (85.139(17)°) and Hg1-Br2-Hg1ⅰ (94.859(17)°) shows a little distortion in center of molecule. This distortion must be attributed to the use of Hg(Ⅱ) orbitals with high s orbital character for bonding to ylidic carbon and the steric effects of phosphine group causing the C1-Hg-Br2 angle to be larger. Since the stabilized resonance structure for the parent ylides is destroyed by the complex formation, P1-C1 bond length in complex C2 (0.178 2(2) nm) is significantly longer than the corres-ponding distances found in the similar non-complexed phosphorus ylides[29-30]. One interesting point in this crystal is unusual hydrogen bonds like C28-H28…Br1ⅰ (H28…Br1ⅰ 0.280 nm) and C17-H17…O1 (H17…O1 0.237 nm) that all together stabilizes the crystal packing.
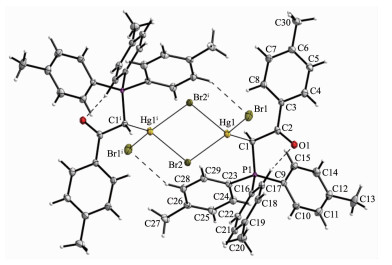
Displacement ellipsoids are drawn at the 50% probability level; Dashed line: C-H…O contacts; Symmetry codes: ⅰ-x, -y+1, -z
下载:
导出CSV
| Experimental | Calculated | |
| Hg1-C1 | 0.226 6(2) | 0.226 587 |
| Hg1-Br1 | 0.254 77(5) | 0.254 763 |
| Hg1-Br2 | 0.267 49(5) | 0.267 495 |
| Hg1-Br2ⅰ | 0.285 32(5) | 0.285 328 |
| P1-C1 | 0.178 2(2) | 0.178 215 |
| O1-C2 | 0.123 1(3) | 0.123 015 |
| C1-C2 | 0.148 9(3) | 0.148 953 |
| C1-Hg1-Br1 | 116.68(6) | 116.68253 |
| C1-Hg1-Br2 | 126.06(6) | 126.05168 |
| Br1-Hg1-Br2 | 108.250(16) | 108.25162 |
| C1-Hg1-Br2ⅰ | 98.91(6) | 98.90585 |
| Br1-Hg1-Br2ⅰ | 117.084(12) | 117.08083 |
| Br2-Hg1-Br2ⅰ | 85.139(17) | 85.13681 |
| Hg1-Br2-Hg1ⅰ | 94.859(17) | 94.86319 |
| C1-P1-C9 | 112.67(11) | 112.68022 |
| C2-C1-P1 | 111.56(16) | 111.53454 |
| Symmetry codes: ⅰ -x, -y+1, -z | ||
下载:
导出CSV
| D-H…A | Method | d(D-H) / nm | d(H…A) / nm | d(D…A) / nm | ∠DHA / (°) |
| C17-H17…O1 | experimental | 0.095 | 0.237 | 0.311 8 | 136 |
| calculated | 0.095 | 0.237 | 0.318 3 | 135.6 | |
| C28-H28…Br1ⅰ | experimental | 0.095 | 0.280 | 0.367 7 | 154 |
| calculated | 0.095 | 0.280 | 0.367 7 | 153.5 | |
| C27-H27…O1ⅱ | 0.098 | 0.256 | 0.316 4 | 120 | |
| C5-H5…Cg1ⅲ | 0.095 | 0.278 | 0.367 5 | 157 | |
| C7-H7…Cg2ⅳ | 0.095 | 0.272 | 0.361 7 | 158 | |
| C25-H25…Cg3ⅴ | 0.095 | 0.271 | 0.345 1 | 136 | |
| Symmetry codes: ⅰ -x, -y+1, -z; ⅱ x, -y+1/2, z-1/2; ⅲ x+1, y+1/2, z+1/2; ⅳ x, y+1, z; ⅴ x+1, -y+1, -z; Cg1, Cg2, Cg3 are the centroids of C9~C15, C16~C22 and C3~C8 ring, respectively. | |||||
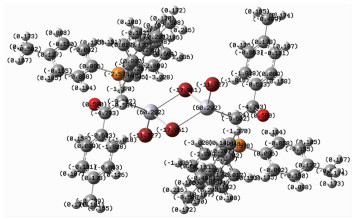
The single point (SP) structure of complex C2 calculated at B3LYP/6-31G* level of DFT method is showed in Fig. 3. It should be mentioned that the corresponding distances in SP structure are equal to the experimental values, due to the freezing of non-hydrogen atoms in the research. According to the results, the total energy and dipole moment of complex C2 is -439 099.94 eV and 0.000 0 Debye which seems to be thermodynamically most stable structure (Fig. 2). Since complex C2 has a crystalline nature and its X-ray diffraction data are available and due to the fact that the X-ray is not able to identify the position of hydrogen atoms positions, all atoms except hydrogen atoms in this research were frozen. The SP calculation was just performed to obtain the correct position of hydrogen atoms.
Moreover, determination of the stable configura-tions of single complex C2 was accomplished regarding the molecular electrostatic potential (MEP) plot of single complex C2 which is represented in Fig. 4. As shown in Fig. 4, the more partial positive charge around the Hg atoms in structure are reactive sites toward interactions with other ylides or Schiff bases which are powerful than ylide L. The possibility of a strong ligand molecule approaching the outer surface of the complex is provided, then ligand L and its bonds to Hg atoms is removed. The obtained structural results by experimental methods confirmed the MEP analysis. This finding will help us to increase our knowledge about formed interactions nature between the studied complex and ligands.
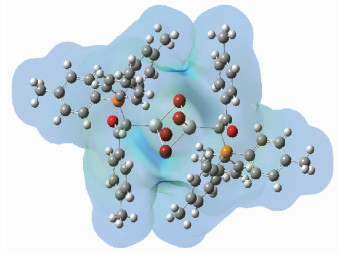
Color range: blue, more positive than 0.441 a.u.; green, 0.441~0 a.u.; yellow, 0~-0.441 a.u.; red, less than-0.441 a.u.
The surfaces are defined by the 0.000 4 electrons/b3 contour of electronic density. Furthermore, since the charge distribution on the structure plays an important role in creating inter and intra-molecular interactions, it was attempted to calculate the quantitative values of atomic charges (Fig. 5). Based on the results, the charge on Hg atoms is about 60.292 showing very low electron density caused by the adjacent bromide atoms which attract the electrons of mercury. The negative charge of bridged bromide is -17.961 and for terminal bromide is -11.727, which demonstrates that terminal bonds are polar than bridged bonds. So, terminal bonds are expected to be stronger than bridged bonds, which is confirmed by X-ray data.
Comparison between the experimental and calculated bond lengths and bond angles for complex C2 is presented in Table 2. The calculated structure of C2 in the gas-phase is in accordance with the structure observed by X-ray crystallography. A comparison between the experimental and calculated hydrogen bonds for complex C2 shows that the calculated structure of C2 in the gas-phase resembles the structure observed by X-ray crystallography (Table 3).
The present study describes the coordination chemistry and spectral characterization of mercury(Ⅱ) and cadmium(Ⅱ) complexes with ylide ligand(C1 and C2). On the basis of spectroscopic data, it is proposed that L herein exhibits monodentate C-coordination to the metal center. In addition, X-ray crystallography studies confirm the trans-like dimeric structure for complex C2 with L. Computational studies at B3LYP/6-31G* level of DFT theory were used to appoint C2 reactive sites.
Bhatti R S, Shah S, Suresh, et al. Int. J. Med. Chem., 2013:793260
Heng S, Tieu W, Hautmann S, et al. J. Bioorg. Med. Chem., 2011, 19:7453-7463 doi: 10.1016/j.bmc.2011.10.042
Arimori S, Matsubara O, Takada M, et al. R. Soc. Open Sci., 2016, 3(5):UNSP160102 doi: 10.1098/rsos.160102
Spengler G, Ocsovszki I, Tönki ÁS, et al. Anticancer Res., 2015, 35:5915-5919
Karami K, Hosseini-Kharat M, Shirani-Sarmazeh Z, et al. J. Coord. Chem., 2016, 69(5):763-778 doi: 10.1080/00958972.2016.1151876
Maigali S S, Abd-El-Maksoud M A, El-Hussieny M, et al. J. Chem. Res., 2014, 38:754-761 doi: 10.3184/174751914X14179446629855
Sabounchei S J, Panahimehr M, Khavasi H R, et al. Chem. Pap., 2014, 68:624-632
Li X Y, Stephan J, Harms K, et al. Organometallics, 2004, 23:3359-3361 doi: 10.1021/om049742h
Sabounchei S J, Ahmadi M, Nasri Z, et al. Tetrahedron Lett., 2013, 54:4656-4660 doi: 10.1016/j.tetlet.2013.06.064
Brownie J H, Baird M C. Coord. Chem. Rev., 2008, 252:1734-1754 doi: 10.1016/j.ccr.2007.12.028
Sabounchei S J, Dadrass A R, Nemattalab H. Asian J. Chem., 2008, 20:4329-4334
Sabounchei S J, Samiee S, Salehzadeh S, et al. J. Organomet. Chem., 2010, 695:1441-1450 doi: 10.1016/j.jorganchem.2010.02.029
Sabounchei S J, Zamanian M, Pourshahbaz M, et al. J. Chem. Res., 2016, 40:130-136 doi: 10.3184/174751916X14537279217786
CrysAlisPro, Agilent Technologies, Yarnton, Oxfordshire, England, 2012.
Sabounchei S J, Nemattalab H, Salehzadeh S, et al. J. Organomet. Chem., 2008, 693:1975-1985 doi: 10.1016/j.jorganchem.2008.02.025
Groom C R, Allen F H. Angew. Chem. Int. Ed., 2014, 53:662-671 doi: 10.1002/anie.201306438
Sheldrick G M. Acta Crystallogr. Sect. A:Found. Crystallogr., 2008, 64:112-122 doi: 10.1107/S0108767307043930
Brandenburg K. Diamond Vers. 3.2k, Bonn, Germany, 2014.
Labanowski J K, Andzelm J W. Density Functional Methods in Chemistry. New York:Springer, 1991:443
Schmidt M W, Baldridge K K, Boatz J A, et al. J. Comput. Chem., 1993, 11:1347-1363
Lee C T, Yang W T, Parr R G. Phys. Rev. B:Condens. Matter, 1988, 37:785-789 doi: 10.1103/PhysRevB.37.785
Yamada J, Hashimoto H, Inomata Y, et al. Bull. Chem. Soc. Jpn., 1994, 67:3224-3230 doi: 10.1246/bcsj.67.3224
Cowan S W, Dakternieks D, Gable R W, et al. Aust. J. Chem., 1986, 39:547-556 doi: 10.1071/CH9860547
Kalyanasundari M, Panchanatheswaran K, Robinson W T, et al. J. Organomet. Chem., 1995, 491:103-109 doi: 10.1016/0022-328X(94)05217-Y
Sabounchei S J, Nemattalab H, Salehzadeh S, et al. Inorg. Chim. Acta, 2009, 362:105-112 doi: 10.1016/j.ica.2008.03.015
Spencer E C, Mariyatra M B, Howard J A K, et al. J. Organomet. Chem., 2007, 692:1081-1086 doi: 10.1016/j.jorganchem.2006.11.002
Sabounchei S J, Salehzadeh S, Hosseinzadeh M, et al. Polyhedron, 2011, 30:2486-2492 doi: 10.1016/j.poly.2011.06.033
Sabounchei S J, Dadrass A R, Jafarzadeh M, et al. J. Organomet. Chem., 2007, 692:2500-2507 doi: 10.1016/j.jorganchem.2007.02.027
Spencer E C, Kalyanasundari B, Mariyatra M B, et al. Inorg. Chim. Acta, 2006, 359:35-43 doi: 10.1016/j.ica.2005.09.006
Dadrass A R, Ramazani A, Marjani A P, et al. Chin. J. Struct. Chem., 2015, 34(3):373-378

Figure 2 X-ray structure of C2 showing atom-numbering scheme
Displacement ellipsoids are drawn at the 50% probability level; Dashed line: C-H…O contacts; Symmetry codes: ⅰ-x, -y+1, -z

Figure 4 Calculated electrostatic potentials on the molecular surfaces of a single C2 molecule
Color range: blue, more positive than 0.441 a.u.; green, 0.441~0 a.u.; yellow, 0~-0.441 a.u.; red, less than-0.441 a.u.
Table 1. Crystal data and structure refinement for complex C2
| Chemical formula | C60H58Br4Hg2O2P2 | Z | 2 | |
| Mr | 1 593.82 | Crystal size / mm | 0.30×0.27×0.21 | |
| Crystal system | Monoclinic | μ / mm-1 | 8.31 | |
| Space group | P21/c | Measured, independent, observed [I>2σ(I)] reflection | 21 419, 8 434, 7 453 | |
| a / nm | 1.178 9(2) | Rint | 0.022 | |
| b / nm | 1.078 3(2) | R1, wR2 | 0.023, 0.046 | |
| c / nm | 2.244 7(4) | S | 1.10 | |
| β / (°) | 95.63(2) | Parameter | 320 | |
| V / nm3 | 2.839 7(9) | Restraint | 0 | |
| Dc / (g·cm-3) | 1.864 | (Δρ)max, (Δρ)min / (e·nm-3) | 690, -650 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Comparison between experimental and calculated values of bond lengths (nm) and bond angles (°) for complex C2
| Experimental | Calculated | |
| Hg1-C1 | 0.226 6(2) | 0.226 587 |
| Hg1-Br1 | 0.254 77(5) | 0.254 763 |
| Hg1-Br2 | 0.267 49(5) | 0.267 495 |
| Hg1-Br2ⅰ | 0.285 32(5) | 0.285 328 |
| P1-C1 | 0.178 2(2) | 0.178 215 |
| O1-C2 | 0.123 1(3) | 0.123 015 |
| C1-C2 | 0.148 9(3) | 0.148 953 |
| C1-Hg1-Br1 | 116.68(6) | 116.68253 |
| C1-Hg1-Br2 | 126.06(6) | 126.05168 |
| Br1-Hg1-Br2 | 108.250(16) | 108.25162 |
| C1-Hg1-Br2ⅰ | 98.91(6) | 98.90585 |
| Br1-Hg1-Br2ⅰ | 117.084(12) | 117.08083 |
| Br2-Hg1-Br2ⅰ | 85.139(17) | 85.13681 |
| Hg1-Br2-Hg1ⅰ | 94.859(17) | 94.86319 |
| C1-P1-C9 | 112.67(11) | 112.68022 |
| C2-C1-P1 | 111.56(16) | 111.53454 |
| Symmetry codes: ⅰ -x, -y+1, -z | ||
下载: 导出CSV
Table 3. Comparison between experimental and calculated values of hydrogen bond parameters for complex C2
| D-H…A | Method | d(D-H) / nm | d(H…A) / nm | d(D…A) / nm | ∠DHA / (°) |
| C17-H17…O1 | experimental | 0.095 | 0.237 | 0.311 8 | 136 |
| calculated | 0.095 | 0.237 | 0.318 3 | 135.6 | |
| C28-H28…Br1ⅰ | experimental | 0.095 | 0.280 | 0.367 7 | 154 |
| calculated | 0.095 | 0.280 | 0.367 7 | 153.5 | |
| C27-H27…O1ⅱ | 0.098 | 0.256 | 0.316 4 | 120 | |
| C5-H5…Cg1ⅲ | 0.095 | 0.278 | 0.367 5 | 157 | |
| C7-H7…Cg2ⅳ | 0.095 | 0.272 | 0.361 7 | 158 | |
| C25-H25…Cg3ⅴ | 0.095 | 0.271 | 0.345 1 | 136 | |
| Symmetry codes: ⅰ -x, -y+1, -z; ⅱ x, -y+1/2, z-1/2; ⅲ x+1, y+1/2, z+1/2; ⅳ x, y+1, z; ⅴ x+1, -y+1, -z; Cg1, Cg2, Cg3 are the centroids of C9~C15, C16~C22 and C3~C8 ring, respectively. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们







 下载:
下载: