Table 1.
Crystal data and structural refinement parameters for the title complexes 1~3
Citation:
CHEN Xiao-Li, CUI Hua-Li, YANG Hua, REN Yi-Xia, WANG Ji-Jiang, WANG Xiao. Syntheses, Crystal Structures and Properties of Three Metal Complexes Based on 3, 3', 4, 4'-Tetracarboxyazobenzene[J]. Chinese Journal of Inorganic Chemistry,
2018, 34(12): 2298-2306.
doi:
10.11862/CJIC.2018.266

基于3,3′,4,4′-四羧基偶氮苯构筑的三个金属配合物的合成、晶体结构及性质
摘要:
在水热条件下利用H4ddb配体合成了3个过渡金属配合物[Co2(ddb)(phen)2(H2O)6]·3H2O(1),[Co(ddb)0.5(bpy)0.5(H2O)3]n(2)和{[Ag(dpe)]·0.5(H2ddb)·H2O}n(3)(H4ddb=3,3',4,4'-四羧基偶氮苯,bpy=4,4'-联吡啶,dpe=1,2-二(4-吡啶基乙烯)),并用元素分析、红外光谱、X射线粉末衍射、X射线单晶衍射对其进行了表征。配合物1为双核结构,基于丰富的氢键作用扩展形成三维超分子网结构。配合物2为基于钴离子通过ddb4-配体以μ4:η1,η1,η1,η1的配位模式连接而成的二维网结构。配合物3是由Ag(Ⅰ)离子与dpe配体形成的直链结构,客体分子H2ddb2-通过氢键作用将其扩展为三维超分子结构。此外还研究了配合物1~3的荧光性质和热稳定性。
-
关键词:
- 过渡金属
- / 3, 3', 4, 4'-四羧基偶氮苯
- / 晶体结构
- / 荧光
English
Syntheses, Crystal Structures and Properties of Three Metal Complexes Based on 3, 3', 4, 4'-Tetracarboxyazobenzene
Abstract:
Three transition metal complexes based on H4ddb ligand, namely[Co2(ddb)(phen)2(H2O)6]·3H2O (1), [Co(ddb)0.5(bpy)0.5(H2O)3]n (2) and {[Ag(dpe)]·0.5(H2ddb)·H2O}n (3). (H4ddb=3, 3', 4, 4'-tetracarboxyazobenzene, bpy=4, 4'-pyridine, dpe=1, 2-di(4-pyridyl) ethylene) have been synthesized and structurally characterized by elemental anal-yses, IR spectroscopy, powder X-ray diffraction and single-crystal X-ray diffraction analyses. Complex 1 is binuclear structure, which is linked into 3D supramolecular network through rich hydrogen bonding interactions. Complex 2 shows 2D network constructed from Co2+ ion cross-linked by ddb4- ligands via μ4:η1, η1, η1, η1 coordina-tion mode. Complex 3 is linear chain structure based on Ag(Ⅰ) ion and dpe ligands. Interestingly, the guest molecule H2ddb2- extends a 3D supramolecular structure of 3 through intermolecular hydrogen bonding interactions. In addition, the thermal stabilities and luminescence properties of 1~3 were also studied.
-
Key words:
- transition metal
- / 3, 3', 4, 4'-tetracarboxyazobenzene
- / crystal structure
- / luminescence
-
0. Introduction
The crystal engineering of metal-organic frame-works (MOFs) have been attracted extensive attention, not only because of their fantastic topological structures but also promising properties in luminescence, magne-tism, catalysis, gas absorption and separation and so on[1-7]. Although a variety of metal MOFs with desired structures and functions have been synthesized to date, rational control in the construction of polymers remains a great challenge in crystal engineering. In order to prepared MOFs with diverse structures and desired functionalities, judicious selection of approp-riate polydentate organic ligands and metal ions is one of the most efficient strategies[8-11]. So many polycar-boxylate ligands are often employed as bridging ligands to construct MOFs, due to their extension ability both in covalent bonding and in supramolecular interac-tions (H-bonding and aromatic stacking)[12-15].
As a member of polycarboxylate ligands, 3, 3′, 4, 4′-tetracarboxyazobenzene (H4ddb) has four carboxyl groups that may be completely or partially deproto-nated, and can provide hydrogen bond donors and acceptors, which makes it a wonderful candidate for the construction of supramolecular networks depending upon the number of deprotonated carboxylate groups. Therefore, H4ddb may be an excellent candidate for the construction of multidimensional coordination polymers.
However, to the best of our knowledge, ddb-metal complex have rarely been reported[16-21], and much work is still necessary to understand the coordination chem-istry of ddb4- ligand. We also notice that the introduc-tion of N-containing auxiliary ligands such as 1, 10-phenanthroline (phen), 4, 4′-bipyridine (bpy), 1, 2-bis(4-pyridyl)ethane (bpe) or 1, 2-di(4-pyridyl)ethylene (dpe) via adjustment of the carboxylate bridging mode into the system may lead to new structural evolution and fine-tuning the structural motif of the complexes[22-25]. With the aim of understanding the coordination chemistry of them and studying the influence on the framework structure of the complexes, we have recently engaged in the research of this kind of complex. Luckily, we have now obtained three complexes, [Co2(ddb)(phen)2(H2O)6]·3H2O (1), [Co(ddb)0.5(bpy)0.5(H2O)3]n (2) and {[Ag(dpe)]·0.5(H2ddb)·H2O}n (3). Herein we reported their syntheses, structures, thermal stabilities and luminescent properties.
1. Experimental
1.1 Reagents and physical measurements
All chemicals and reagents were used as received from commercial sources without further purification. All reactions were carried out under hydrothermal conditions. Elemental analyses (C, H, N) were determined with a Elementar Vario EL Ⅲ elemental analyzer. IR spectra were recorded as KBr pellets on a Bruker EQUINOX55 spectrophotometer in the 4 000~400 cm-1 region. Fluorescence spectra were performed on a Hitachi F-4500 fluorescence spectro-photometer at room temperature. Thermogravimetric analyses (TGA) were performed in a nitrogen atmo-sphere with a heating rate of 10 ℃·min-1 with a NETZSCHSTA 449C thermogravimetric analyzer. The X-ray powder diffraction pattern (XRD) was recorded with a Rigaku D/Max Ⅲ diffractometer operating at 40 kV and 30 mA using Mo Kα radiation (λ=0.154 18 nm) in the range of 5°~50°.
1.2 Synthesis of [Co2(ddb)(phen)2 (H2O)6]·3H2O (1)
A mixture of Co(Ac)2·4H2O (24.9 mg, 0.1 mmol), H4ddb (35.8 mg, 0.1 mmol), phen (19.8 mg, 0.1 mmol) and water (10 mL) was stirred and adjusted to pH 6.5 with 0.5 mol·L-1 NaOH solution, then sealed in a 25 mL Telfon-lined stainless steel container, which was heated to 160 ℃ for 96 h. Then cooling to room temperature at a rate of 5 ℃·h-1. Brown crystals were obtained in ca. 53% yield based on Co. Anal. Calcd. for C40H40Co2N6O17(%): C, 48.30; H, 4.05; N, 8.45. Found(%): C, 48.49; H, 3.72; N, 8.45. FI-IR (KBr, cm-1): 3 394(s), 3 072(s), 1 628(m), 1 553(s), 1 486(m), 1 422(s), 1 209(w), 1 140(w), 1 070(w), 922(w), 845(m), 801(m), 726(m), 671(w).
1.3 Synthesis of [Co(ddb)0.5(bpy)0.5(H2O)3]n (2)
The brown crystals of 2 were prepared by a similar method used in the synthesis of 1 except that phen was replaced by bpy (Yield: 45% based on Co). Anal. Calcd. for C13H13CoN2O7(%): C, 42.41; H, 3.56; N, 7.61. Found(%): C, 42.43; H, 3.52; N, 7.63. FI-IR (KBr, cm-1): 3 387(s), 3 090(s), 1 610(m), 1 564(s), 1 473(m), 1 409(s), 1 202(w), 1 056(w), 1 102(w), 906(w), 845(m), 804(m), 720(m), 678(w).
1.4 Synthesis of {[Ag(dpe)]·0.5(H2ddb)·H2O}n (3)
The colorless crystals of 3 were prepared by a similar method used in the synthesis of 1 except that phen was replaced by dpe and Co(Ac)2·4H2O (24.9 mg, 0.1 mmol) was replaced by AgNO3 (16.9 mg, 0.1 mmol) (Yield: 39% based on Ag). Anal. Calcd. for C20H16AgN3O5(%): C, 49.61; H, 2.91; N, 8.68. Found(%): C, 49.67; H, 2.85; N, 8.65. FI-IR (KBr, cm-1): 3 429(s), 3 037(w), 1 701(s), 1 607(s), 1 495(s), 1 357(s), 1 240(w), 1 202(m), 1 068(w), 969(m), 831(m), 771(w), 617(w), 547(m).
1.5 X-ray crystallography
Intensity data were collected on a Bruker Smart APEX Ⅱ CCD diffractometer with graphite-monochro-mated Mo Kα radiation (λ=0.071 073 nm) at room temperature. Empirical absorption corrections were applied using the SADABS program[26a]. The structures were solved by direct methods and refined by the full-matrix least-squares based on F2 using SHELXTL-97 program[26b]. In 3, one dpe molecule and two oxygen atoms were split into two site with an occupancy ratio 0.5:0.5 for C6/C6A, C7/C7A, O3/O3A and O6/O6A. All non-hydrogen atoms were refined anisotropically and hydrogen atoms of organic ligands were generated geometrically. Crystal data and structural refinement parameters for 1~3 are summarized in Table 1, selected bond distances and bond angles are listed in Table 2.
Table 1

 下载:
导出CSV
下载:
导出CSV
Complex 1 2 3 Empirical formula C40H40Co2N6O17 C13H13CoN2O7 C20H16AgN3O5 Formula weight 994.64 368.18 486.23 Crystal system Monoclinic Triclinic Triclinic Space group C2/c P1 P1 a / nm 2.179 4(2) 0.598 02(5) 0.736 92(11) b / nm 0.800 25(9) 0.874 66(7 1.143 28(17) c / nm 2.595 4(3) 1.418 60(11) 1.243 91(19) α / (°) 101.296 0(10) 75.985(2) β / (°) 108.050(2) 92.210 0(10) 82.837(2) γ / (°) 100.118 0(10) 79.311(2) V / nm3 4.303 8(8) 0.714 28(10) 0.995 7(3) Z 4 2 2 Dc / (g·cm-3) 1.53 1.712 1.622 θ range for data / (°) 1.65~28.43 1.47~25.50 1.69~25.50 Absorption coefficient / mm-1 0.853 1.242 1.049 Crystal size / mm 0.34×0.29×0.26 0.30×0.28×0.27 0.31×0.27×0.25 F(000) 2 036 376 488 Reflection collected 12 972 3 731 5 162 Unique reflection (Rint) 5 266 (0.039 2) 2 636 (0.009 7) 3 610 (0.012 5) Observed reflection [I>2σ(I)] 3 582 2 533 3 149 Limiting-indices -22 ≤ h ≤ 29,
-10 ≤ k ≤10,
-34 ≤ l ≤ 34-6 ≤ h ≤ 7,
-10 ≤ k ≤10,
-17 ≤ l ≤ 14-8 ≤ h ≤ 7,
-13 ≤ k ≤9,
-14 ≤ l ≤ 15Goodness-of-fit (on F2) 1.098 1.198 1.107 R1, wR2 [I>2σ(I)] 0.051 7, 0.113 7 0.024 2, 0.072 2 0.042 9, 0.107 9 R1, wR2 (all data) 0.090 7, 0.140 3 0.026 9, 0.084 2 0.050 0, 0.111 8 Table 2
Table 2. Selected bond distances (nm) and bond angles (°) for complexes 1~3下载:
导出CSV
Complex 1 Co(1)-O(1) 0.207 2(2) Co(1)-O(6) 0.209 8(2) Co(1)-O(5) 0.210 1(3) Co(1)-O(7) 0.212 9(2) Co(1)-N(3) 0.212 0(3) Co(1)-N(2) 0.215 5(3) O(1)-Co(1)-O(6) 95.52(9) O(1)-Co(1)-O(5) 90.68(10) O(5)-Co(1)-N(3) 94.69(10) O(1)-Co(1)-O(7) 92.74(9) O(1)-Co(1)-N(2) 167.25(10) O(6)-Co(1)-N(2) 97.22(10) O(6)-Co(1)-O(5) 90.99(10) O(6)-Co(1)-O(7) 83.86(9) O(5)-Co(1)-N(2) 89.17(11) O(1)-Co(1)-N(3) 89.87(9) O(5)-Co(1)-O(7) 174.07(10) N(3)-Co(1)-N(2) 77.44(11) O(6)-Co(1)-N(3) 172.12(10) N(3)-Co(1)-O(7) 90.17(10) O(7)-Co(1)-N(2) 88.56(10) Complex 2 Co(1)-O(1) 0.201 92(15) Co(1)-O(7) 0.206 55(17) Co(1)-O(5) 0.208 18(16) Co(1)-O(6) 0.209 25(15) Co(1)-N(2) 0.217 18(17) Co(1)-O(3A) 0.219 89(13) O(1)-Co(1)-O(7) 173.42(7) O(1)-Co(1)-O(5) 86.64(7) O(7)-Co(1)-O(5) 88.93(7) O(1)-Co(1)-O(6) 93.77(7) O(7)-Co(1)-O(6) 90.77(7) O(5)-Co(1)-O(6) 178.62(6) O(1)-Co(1)-N(2) 90.10(7) O(7)-Co(1)-N(2) 85.04(7) O(5)-Co(1)-N(2) 90.04(6) O(6)-Co(1)-N(2) 91.28(6) O(1)-Co(1)-O(3A) 95.47(6) O(7)-Co(1)-O(3A) 89.52(7) O(5)-Co(1)-O(3A) 91.73(6) O(6)-Co(1)-O(3A) 86.92(6) N(2)-Co(1)-O(3A) 174.25(6) Complex 3 Ag(1)-N(1) 0.215 3(3) Ag(1)-N(2) 0.215 5(3) N(1)-Ag(1)-N(2) 172.02(12) Symmetry codes: A: 2-x, -y, -z; B: 2-x, -y, 1-z for 2. CCDC: 1861508, 1; 1861509, 2; 1861510, 3.
2. Results and discussion
2.1 Structure description of complex 1
Single-crystal X-ray diffraction analysis reveals that complex 1 is a binuclear structure crystallizing in monoclinic system with C2/c space group. The asymmetric unit of 1 contains one independent Co(Ⅱ)ion, half ddb4- ligand, one coordinated phen ligand, three coordinated water molecules and one and a half lattice water molecules. As shown in Fig. 1, Co1 is surrounded by two nitrogen atoms (N2, N3) from one chelating phen ligand, one oxygen atom (O1) from one bridging carboxylate groups of ddb4- ligand, three oxygen atoms from three coordinated water molecule. The Co1-O distances fall in the range of 0.207 2(2)~0.212 9(2) nm and are similar to those found in other cobalt carboxylate complexes[27]. The coordination geometry of the Co1 center can be described as a distorted octahedral geometry.
Figure 1
 Figure 1. Coordination environment of Co(Ⅱ) ion in 1
Figure 1. Coordination environment of Co(Ⅱ) ion in 1All H atoms are omitted for clarity; Symmetry codes: A:1-x, -1-y, 1.5-z
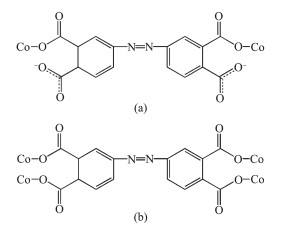
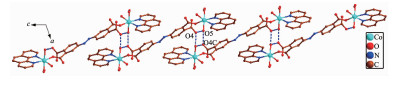
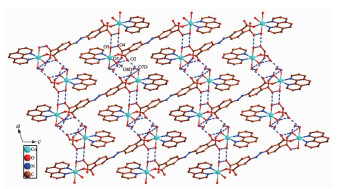
In 1, H4ddb is completely deprotonated and adopts a μ2:η1, η0, η1, η0 coordination mode (Scheme 1a). Two carboxylate groups coordinate with two Co(Ⅱ) ions monodentately. Although ddb4- ion participate in the coordination, two benzene rings of ddb4- ion have not been distorted, and the dihedral angle between the two phenyl rings is 0° for 1. On the basis of the connection mode, each pair of Co(Ⅱ) ions are bridged by two carboxylate groups from one ddb4- ligand to form a binuclear structure with the Co1…Co1 distances of 1.626 8 nm. The binuclear structure are linked by the hydrogen bonding interactions (O5…O4B, 0.284 8(3) nm; O6…O3B, 0.269 6 nm) generating a 1D double-chain (Fig. 2). The adjacent double-chain recognizes each other to generate a 2D bilayer supra-molecular network via hydrogen bonding interactions (O5…O4, 0.284 8(3) nm; O5…O4C, 0.277 5 nm), which is further developed into 3D supramolecular structure by hydrogen bonding interactions (O7…O2 0.275 5 nm, O6…O2 0.264 3(3) nm, O7…O8 0.279 1(6) nm, O7…O8A 0.258 7(8) nm, O8…O3 0.272 9 nm, O9…O3 0.283 0 nm, Fig. 3).
Scheme 1
Figure 2
 Figure 2. View of 1D double-chain of 1 formed by hydrogen bonding interactions
Figure 2. View of 1D double-chain of 1 formed by hydrogen bonding interactionsAll H atoms are omitted for clarity; Symmetry codes: B: x, -1+y, z
Figure 3
 Figure 3. View of 2D bilayer supramolecular network of 1 via hydrogen bonding interactions along b-axis
Figure 3. View of 2D bilayer supramolecular network of 1 via hydrogen bonding interactions along b-axisAll H atoms are omitted for clarity; Symmetry codes: C: 0.5-x, 1.5+y, 0.5-z
2.2 Structure description of complex 2
To further examine the influence of the auxiliary ligands on the structure of 1, a longer bridge ligand bpy is used instead of phen. Consequently, a novel 2D polymeric network was obtained. The asymmetric unit of 2 contains one Co(Ⅱ) atom, a half ddb4- ligand, a half bpy ligand and three coordinated water molecules (Fig. 5). Each Co1 atom is six coordinated by one nitrogen atom (Co1-N2 0.217 82(17) nm) from one bpy ligand, two carboxylate group oxygen atoms from two ddb4- ligands and three water molecules. The bond lengths of Co-O are comparable to the published ones[28], varying between 0.201 92(15) and 0.219 89(13) nm. The coordination geometry of Co1 center can be described as a distorted octahedral geometry.
Figure 4
 Figure 4. View of 3D supramolecular architecture of 1 based on hydrogen bonding interaction along b-axis
Figure 4. View of 3D supramolecular architecture of 1 based on hydrogen bonding interaction along b-axisAll H atoms are omitted for clarity; Symmetry codes: D: 2-x, 3-y, -z
Figure 5
 Figure 5. Coordination environment of Co(Ⅱ) ion in 2
Figure 5. Coordination environment of Co(Ⅱ) ion in 2All H atoms are omitted for clarity; Symmetry codes: A: 2-x, -y, -z, B: 2-x, -y, 1-z
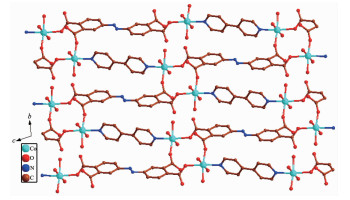
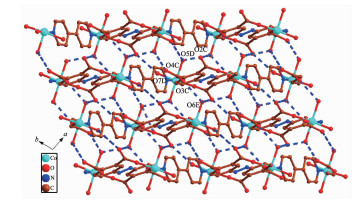
In 2, H4ddb are completely deprotonated and adopts a μ4:η1, η1, η1, η1 coordination mode (Scheme 1b). Four carboxylate groups adopt a bridging monodentate coordination mode connecting four Co(Ⅱ) ions (Scheme 1b). Based on the connection mode, a pair of Co(Ⅱ) ions are bridged by four carboxylate oxygen atoms from two ddb4- ions to form a 14-membered ring {Co2O4C8} (ring a) with the Co1…Co1 distance of 0.646 1 nm. Meanwhile, four Co(Ⅱ) ions are also bridged by two ddb4- ions and two bpy ligands to form a 46-membered ring {Co4O4N8C30} (ring b) containing a type of pore with size of ca. 2.481 8 nm×2.520 8 nm based on the distances of Co1…Co1 and C1…C1. Interes-tingly, these 14-member and 46-membered rings were arranged alternately to form a 2D network (Fig. 6). Two adjacent 2D networks are packed into 3D supramole-cular structure by the hydrogen bonding interactions (O5D…O4C 0.266 3 nm, O7D…O2C 0.289 4 nm, O6E…O3C 0.274 3 nm, O6E…O2 0.259 3 nm, O5D…O6E 0.284 6 nm, Fig. 7).
Figure 6
Figure 7
 Figure 7. View of 3D supramolecular architecture based on hydrogen bonding interaction along c-axis
Figure 7. View of 3D supramolecular architecture based on hydrogen bonding interaction along c-axisAll H atoms are omitted for clarity; Symmetry codes: C: 2-x, 1-y, 3-z, D: x, 1+y, 2+z, E: 1-x, 1-y, 2-z
2.3 Structure description of complex 3
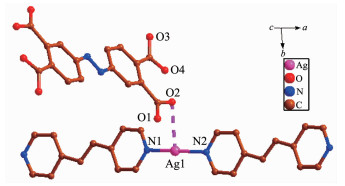
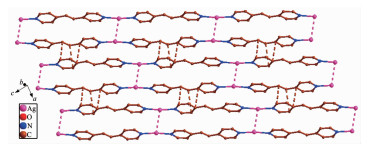
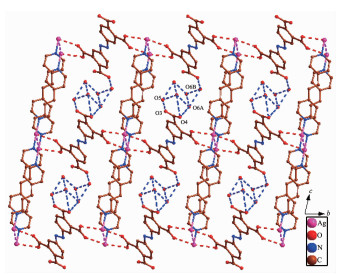
The asymmetric unit of 3 has one independent Ag(Ⅰ) ion, one dpe ligand, a half free H2ddb2- ion and one lattice water molecule (Fig. 8). Ag1 center coor-dinated with two nitrogen atoms (N1, N2) from two different dpe ligands (Ag1-N2 0.215 5(3) nm, Ag1-N1 0.215 3(3) nm) to form a slightly distorted linear geometry. However, the distance of 0.283 9 nm between Ag1-O2 suggests a non-negligible interaction between them. Thus, the coordination polyhedron of Ag(Ⅰ) ion can also be described as a T-shaped coordination geometry. The N1-Ag1-N2 bond angle is 172.02°, and the three atoms almost in a line. The pyridyl rings of dpe ligand are non-coplanar with a dihedral angle of 7.18° and the Ag1…Ag1 separation based on dpe ligand is 1.364 5 nm. Hence, the dpe ligands bridge Ag1 centers to form 1D linear chain. The Ag1…Ag1distance between two parallel linear chains is 0.321 0 nm, which is less than the van der Waals contact whose distance is 0.340 nm, illustrating the existence of argentophilic interactions between Ag(Ⅰ) ions. Based on the argentophilic interactions between Ag(Ⅰ) ions, the adjacent linear chains form a 1D double chain structure. Interestingly, the adjacent double chains interact with each other to generate a 2D supramolecular network through the π…π stacking interactions with an edge-to-edge distance of 0.338 7 nm between two pyridine rings of dpe ligands and 0.337 0, 0.338 8 nm between the pyridine ring of dpe ligand and the double bonds of dpe ligand, respectively (Fig. 9). These kinds of π…π stacking interactions are in an alternate fashion and consolidate the stacked arrangement. The adjacent 2D structures further form a 3D supramolecular structure through O-H…O hydrogen bonds (O6A…O4 0.289 9 nm, O5…O3 0.288 4 nm, O6B…O6A 0.254 7 nm, O5…O6 0.285 0 nm, O6A…O6B 0.270 9 nm) and Ag…O week intera-ctions (Ag1A…O1 0.276 0 nm, Ag1…O2 0.283 9 nm, Fig. 10).
Figure 8
 Figure 8. Coordination environment of Ag(Ⅰ) ion in 3
Figure 8. Coordination environment of Ag(Ⅰ) ion in 3All H atoms are omitted for clarity
Figure 9
 Figure 9. View of 2D supramolecular network of 3 via Ag…Ag interactions and π…π stacking interactions
Figure 9. View of 2D supramolecular network of 3 via Ag…Ag interactions and π…π stacking interactionsFigure 10
 Figure 10. View of 3D supramolecular structure of 3 based on hydrogen bonding interaction and Ag…O week interactions
Figure 10. View of 3D supramolecular structure of 3 based on hydrogen bonding interaction and Ag…O week interactionsAll H atoms are omitted for clarity; Symmetry codes: A: 2-x, 1-y, 1-z, B: 2-x, 1-y, 2-z
2.2 IR Spectra of 1 and 2
In the FT-IR spectra, the absorption bands in the region of 3 387~3 429 cm-1 may attribute to the stretching vibrations of O-H and N-H. The bands in the region 3 037~3 090 cm-1 can be ascribed to C-H stretching vibrations of the benzene ring[29]. The absence of the absorption bands at 1 730~1 690 cm-1 in 1 and 2 indicates the H4ddb ligand adopts the complete deprotonated ddb4- form, which is consistent with the X-ray structural analysis. The bands in the region of 1 578~1 628 cm-1 for 1~3 can be assigned to the N=N stretching vibrations. The asymmetric stretching vibrations of the carboxylate groups(νas) were observed at 1 553, 1 564 and 1 495 cm-1, and the symmetric stretching vibration (νs) of the carboxylate groups were observed at 1 422, 1 409 and 1 357 cm-1, respectively[30]. The separation Δν(COO) between the νas(COO) and νs(COO) band for 1~3 are 131, 155 and 138 cm-1, which is smaller than 200 cm-1, indicating that the carboxyl groups are coordinated in bridging mode[31].
2.3 Luminescent properties
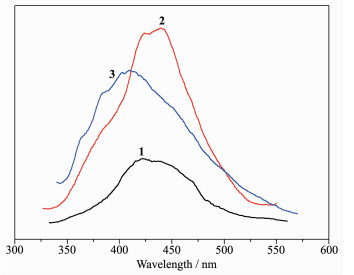
The photoluminescence properties of complexes 1~3 were examined at room temperature, and the emission spectra are shown in Fig. 11. The H4ddb ligand exhibited one very week emission band at 470 nm upon excitation at 294 nm. Upon excitation of solid samples of 1~3 at 284 nm, these complexes showed one emission peak at 421 nm for 1, 438 nm for 2, 410 nm for 3. In comparison with H4ddb ligand, the emission peaks of complexes 1~3 were blue-shifted, which may be due to the π*→π intraligand fluorescence because of close resemblance to the emission band of H4ddb ligand[32]. By comparing the emission spectra of 1~3 and the free ligand, we can conclude that the enhancement of luminescence in 1~3 may be attributed to the ligation of ligand to the metal center, which effectively increases the rigidity and reduces the loss of energy by radiationless decay[33-34].
Figure 11
2.4 Thermal properties and PXRD measurement of complexes 1~3
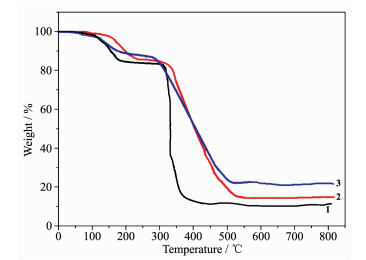
To study the thermal stabilities of these complexes, thermal gravimetric analysis (TGA) were performed. The TG curve of 1~3 are shown in Fig. 12. Complex 1 first lost its coordinated and lattice water molecule below 210 ℃ (Obsd. 14.68%, Calcd. 14.52%). Then 1 was relatively stable up to 210~345 ℃. The second weight loss is 74.49% in the temperature range of 345~400 ℃ corresponding to the decomposition ddb4- and phen ligands (Calcd. 75.23%). Complex 2 first lost its coordinated water molecules below 225 ℃ (Obsd. 14.88%, Calcd. 14.67%). Then 2 was relatively stable up to 225~320 ℃, followed by a continuously two-step weight loss of 73.41% from 320 to 515 ℃ (Calcd. 72.42%), corresponding to the loss of ddb4- and phen ligands. The remaining weight of 12.69% is the final product CoO (Calcd. 12.91%). The TG curve of 3 showed an initial weight loss of 3.69% below 130 ℃ corresponding to the removal of lattice water molecules (Calcd. 3.72%). Then 3 was stable up to 260 ℃ and followed by the weight loss in the range of 260~490 ℃, assigned to the decomposition of ddb4- and dpe ligands (Calcd. 73.18%, Obsd. 72.57%). The remaining weight of 23.68% is Ag2O that is in agreement with the calculated value of 23.76%.
Figure 12
In order to confirm the phase purity of the bulk materials, powder X-ray diffraction (PXRD) patterns were measured at room temperature. The PXRD experimental and computer-simulated patterns of all of them are shown in Fig. 13. The peaks of the simulated and experimental PXRD patterns are in good agreement with each other, confirming the phase purities of 1~3.
Figure 13
3. Conclusions
In summary, we succeeded in getting access to three transition metal complexes based on 3, 3′, 4, 4′-tetracarboxyazobenzene and different auxiliary ligands through hydrothermal method. Complex 1 is binuclear structure. Complex 2 shows 2D network constructed from Co2+ ion cross-linked by ddb4- and bpy ligand. Complex 3 is linear chain structure. Interestingly, the guest molecule ddb4- exists in the structure and extends a 3D supramolecular structure through hydrogen bonding, Ag…Ag and Ag…O interactions. Different structures of complexes 1~3 indicate that the ddb4- ligand has the ability of adjusting its coordination modes and configurations in different reaction systems. Furthermore, the π…π stacking interactions probably play a crucial role to the arrangement and stability of the chain structure, which influence the final supramolecular structures together with abundant hydrogen-bond interactions.
-
-
[1]
Yang X, Xu Q. Cryst. Growth Des., 2017, 17:1450-1455 doi: 10.1021/acs.cgd.7b00166
-
[2]
Kreno L E, Leong K, Farha O K, et al. Chem. Rev., 2012, 112:1105-1125 doi: 10.1021/cr200324t
-
[3]
Gil-Hernández B, Savvin S, Makhloufi G, et al. Inorg. Chem., 2015, 54:1597-1605 doi: 10.1021/ic502586a
-
[4]
Ding L G, Yao B J, Jiang W L, et al. Inorg. Chem., 2017, 56:2337-2344 doi: 10.1021/acs.inorgchem.6b03169
-
[5]
Zhao X L, Sun W Y. CrystEngComm, 2014, 16:3247-3258 doi: 10.1039/c3ce41791c
-
[6]
Pang J, Jiang F, Wu M, et al. Chem. Commun., 2014, 50:2834-2836 doi: 10.1039/c3cc48381a
-
[7]
Zhang Y B, Furukawa H, Yaghi O M, et al. J. Am. Chem. Soc., 2015, 137:2641-2650 doi: 10.1021/ja512311a
-
[8]
Zhao J, Wang Y N, Dong W W, et al. Chem. Commun., 2015, 51:9479-9482 doi: 10.1039/C5CC02043C
-
[9]
Gai Y L, Jiang F L, Hong M C, et al. Cryst. Growth Des., 2014, 14:1010-1017 doi: 10.1021/cg401452p
-
[10]
Li X Z, Li M A, Li Z, et al. Angew. Chem. Int. Ed., 2008, 47:6371-6374 doi: 10.1002/anie.v47:34
-
[11]
Cunha D, Yahia M B, Hall S, et al. Chem. Mater., 2013, 25:2767-2776 doi: 10.1021/cm400798p
-
[12]
Patra R, Titi H M, Goldberg I. CrystEngComm, 2013, 15:2853-2862 doi: 10.1039/c3ce27006h
-
[13]
Pan L, Liu H, Lei X, et al. Angew. Chem. Int. Ed., 2003, 42:542-546 doi: 10.1002/anie.200390156
-
[14]
Habib H A, Sanchiz J, Janiak C. Dalton Trans., 2008:4877-4884
-
[15]
Sun D, Han L L, Yuan S, et al. Cryst. Growth Des., 2013, 13:377-385 doi: 10.1021/cg301573c
-
[16]
Wang C C, Jing H P, Zhang Y Q, et al. Transition Met. Chem., 2015, 40:573-584 doi: 10.1007/s11243-015-9950-1
-
[17]
Wang J, Lu L, Wu W P, et al. Synth. React. Inorg. Met.-Org. Nano-Met. Chem., 2012, 42:25-29 doi: 10.1080/15533174.2011.609223
-
[18]
Wang J, Lu L, Wu W P, et al. J. Chem. Res., 2011, 35:424-427 doi: 10.3184/174751911X13101136526834
-
[19]
Wang J, Lu L, Wu W P, et al. Synth. React. Inorg. Met.-Org. Nano-Met. Chem., 2013, 43:791-794 doi: 10.1080/15533174.2012.749909
-
[20]
Wang J, Lu L, Wu W P, et al. Synth. React. Inorg. Met.-Org. Nano-Met. Chem., 2012, 42:1217-1221 doi: 10.1080/15533174.2011.652278
-
[21]
Lu L, Wang J, Bai J W, et al. Cryst. Res. Technol., 2008, 43:1327-1330 doi: 10.1002/crat.v43:12
-
[22]
Lightfoot P, Snedden A. J. Chem. Soc. Dalton Trans., 1999:3549-3551
-
[23]
Sun C Y, Li L C, Jin L P. Polyhedron, 2006, 25:3017-3024 doi: 10.1016/j.poly.2006.05.009
-
[24]
Lee S W, Kim H J, Lee Y K, et al. Inorg. Chim. Acta, 2003, 353:151-158 doi: 10.1016/S0020-1693(03)00246-9
-
[25]
Ahmad M, Sharma M K, Das R, et al. Cryst. Growth Des., 2012, 12:1571-1578 doi: 10.1021/cg201619x
-
[26]
(a) Sheldrick G M. SADABS, University of Göttingen, Germany, 1996.
(b) Sheldrick G M. SHELXS-97 and SHELXL-97, Program for X-ray Crystal Structure Solution and Refinement, Göttingen University, Germany, 1997. -
[27]
王莉, 康全鹏, 郝静, 等.无机化学学报, 2018, 34(3):525-533 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20180315&journal_id=wjhxxbcnWANG Li, KGAN Quan-Peng, HAO Jing, et al. Chinese J. Inorg. Chem., 2018, 34(3):525-533 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20180315&journal_id=wjhxxbcn
-
[28]
刘继伟.无机化学学报, 2017, 33(4):705-712 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20170425&journal_id=wjhxxbcnLIU Ji-Wei. Chinese J. Inorg. Chem., 2017, 33(4):705-712 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20170425&journal_id=wjhxxbcn
-
[29]
Chen S, Fan R Q, Sun C F, et al. Cryst. Growth Des., 2012, 12:1337-1346 doi: 10.1021/cg201411b
-
[30]
Xia J, Wang H S, Shi W, et al. Inorg. Chem., 2007, 46:3450-3458 doi: 10.1021/ic061620p
-
[31]
黄德如, 汪仁庆.无机和配位化合物的红外和拉曼光谱. Beijing: Chemical Industry Press, 1986: 235Nakamoto K, Translated by HUANG De-Ru, WANG Ren-Qing. Infrared Raman Spectra of Inorganic and Coordination Compounds. Beijing: Chemical Industry Press, 1986: 235
-
[32]
Li D X, Chen M M, Li F L, et al. Inorg. Chem. Commun., 2013, 35:302-306 doi: 10.1016/j.inoche.2013.07.014
-
[33]
Zhang L Y, Liu G F, Zheng S L, et al. Eur. J. Inorg. Chem., 2003:2965-2971
-
[34]
Wang X L, Qin C, Wang E B, et al. Angew. Chem. Int. Ed., 2004, 43:5036-5040 doi: 10.1002/(ISSN)1521-3773
-
[1]
-
Figure 1 Coordination environment of Co(Ⅱ) ion in 1
All H atoms are omitted for clarity; Symmetry codes: A:1-x, -1-y, 1.5-z
Figure 2 View of 1D double-chain of 1 formed by hydrogen bonding interactions
All H atoms are omitted for clarity; Symmetry codes: B: x, -1+y, z
Figure 3 View of 2D bilayer supramolecular network of 1 via hydrogen bonding interactions along b-axis
All H atoms are omitted for clarity; Symmetry codes: C: 0.5-x, 1.5+y, 0.5-z
Figure 4 View of 3D supramolecular architecture of 1 based on hydrogen bonding interaction along b-axis
All H atoms are omitted for clarity; Symmetry codes: D: 2-x, 3-y, -z
Figure 5 Coordination environment of Co(Ⅱ) ion in 2
All H atoms are omitted for clarity; Symmetry codes: A: 2-x, -y, -z, B: 2-x, -y, 1-z
Figure 7 View of 3D supramolecular architecture based on hydrogen bonding interaction along c-axis
All H atoms are omitted for clarity; Symmetry codes: C: 2-x, 1-y, 3-z, D: x, 1+y, 2+z, E: 1-x, 1-y, 2-z
Figure 8 Coordination environment of Ag(Ⅰ) ion in 3
All H atoms are omitted for clarity
Figure 9 View of 2D supramolecular network of 3 via Ag…Ag interactions and π…π stacking interactions
Figure 10 View of 3D supramolecular structure of 3 based on hydrogen bonding interaction and Ag…O week interactions
All H atoms are omitted for clarity; Symmetry codes: A: 2-x, 1-y, 1-z, B: 2-x, 1-y, 2-z
Table 1. Crystal data and structural refinement parameters for the title complexes 1~3
Complex 1 2 3 Empirical formula C40H40Co2N6O17 C13H13CoN2O7 C20H16AgN3O5 Formula weight 994.64 368.18 486.23 Crystal system Monoclinic Triclinic Triclinic Space group C2/c P1 P1 a / nm 2.179 4(2) 0.598 02(5) 0.736 92(11) b / nm 0.800 25(9) 0.874 66(7 1.143 28(17) c / nm 2.595 4(3) 1.418 60(11) 1.243 91(19) α / (°) 101.296 0(10) 75.985(2) β / (°) 108.050(2) 92.210 0(10) 82.837(2) γ / (°) 100.118 0(10) 79.311(2) V / nm3 4.303 8(8) 0.714 28(10) 0.995 7(3) Z 4 2 2 Dc / (g·cm-3) 1.53 1.712 1.622 θ range for data / (°) 1.65~28.43 1.47~25.50 1.69~25.50 Absorption coefficient / mm-1 0.853 1.242 1.049 Crystal size / mm 0.34×0.29×0.26 0.30×0.28×0.27 0.31×0.27×0.25 F(000) 2 036 376 488 Reflection collected 12 972 3 731 5 162 Unique reflection (Rint) 5 266 (0.039 2) 2 636 (0.009 7) 3 610 (0.012 5) Observed reflection [I>2σ(I)] 3 582 2 533 3 149 Limiting-indices -22 ≤ h ≤ 29,
-10 ≤ k ≤10,
-34 ≤ l ≤ 34-6 ≤ h ≤ 7,
-10 ≤ k ≤10,
-17 ≤ l ≤ 14-8 ≤ h ≤ 7,
-13 ≤ k ≤9,
-14 ≤ l ≤ 15Goodness-of-fit (on F2) 1.098 1.198 1.107 R1, wR2 [I>2σ(I)] 0.051 7, 0.113 7 0.024 2, 0.072 2 0.042 9, 0.107 9 R1, wR2 (all data) 0.090 7, 0.140 3 0.026 9, 0.084 2 0.050 0, 0.111 8  下载: 导出CSV
下载: 导出CSV
Table 2. Selected bond distances (nm) and bond angles (°) for complexes 1~3
Complex 1 Co(1)-O(1) 0.207 2(2) Co(1)-O(6) 0.209 8(2) Co(1)-O(5) 0.210 1(3) Co(1)-O(7) 0.212 9(2) Co(1)-N(3) 0.212 0(3) Co(1)-N(2) 0.215 5(3) O(1)-Co(1)-O(6) 95.52(9) O(1)-Co(1)-O(5) 90.68(10) O(5)-Co(1)-N(3) 94.69(10) O(1)-Co(1)-O(7) 92.74(9) O(1)-Co(1)-N(2) 167.25(10) O(6)-Co(1)-N(2) 97.22(10) O(6)-Co(1)-O(5) 90.99(10) O(6)-Co(1)-O(7) 83.86(9) O(5)-Co(1)-N(2) 89.17(11) O(1)-Co(1)-N(3) 89.87(9) O(5)-Co(1)-O(7) 174.07(10) N(3)-Co(1)-N(2) 77.44(11) O(6)-Co(1)-N(3) 172.12(10) N(3)-Co(1)-O(7) 90.17(10) O(7)-Co(1)-N(2) 88.56(10) Complex 2 Co(1)-O(1) 0.201 92(15) Co(1)-O(7) 0.206 55(17) Co(1)-O(5) 0.208 18(16) Co(1)-O(6) 0.209 25(15) Co(1)-N(2) 0.217 18(17) Co(1)-O(3A) 0.219 89(13) O(1)-Co(1)-O(7) 173.42(7) O(1)-Co(1)-O(5) 86.64(7) O(7)-Co(1)-O(5) 88.93(7) O(1)-Co(1)-O(6) 93.77(7) O(7)-Co(1)-O(6) 90.77(7) O(5)-Co(1)-O(6) 178.62(6) O(1)-Co(1)-N(2) 90.10(7) O(7)-Co(1)-N(2) 85.04(7) O(5)-Co(1)-N(2) 90.04(6) O(6)-Co(1)-N(2) 91.28(6) O(1)-Co(1)-O(3A) 95.47(6) O(7)-Co(1)-O(3A) 89.52(7) O(5)-Co(1)-O(3A) 91.73(6) O(6)-Co(1)-O(3A) 86.92(6) N(2)-Co(1)-O(3A) 174.25(6) Complex 3 Ag(1)-N(1) 0.215 3(3) Ag(1)-N(2) 0.215 5(3) N(1)-Ag(1)-N(2) 172.02(12) Symmetry codes: A: 2-x, -y, -z; B: 2-x, -y, 1-z for 2.
下载: 导出CSV
-



















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 4
- 文章访问数: 619
- HTML全文浏览量: 80
 下载:
下载:
