-
[1]
Wu H, Cui Y. Nano Today, 2012, 7(5):414-429 doi: 10.1016/j.nantod.2012.08.004
-
[2]
Lu Z D, Liu N, Lee H W, et al. ACS Nano, 2016, 9(3):2540-2547
-
[3]
Xu J, Wang X F, Wang X W, et al. ChemElectroChem, 2014, 1(6):975-1002 doi: 10.1002/celc.v1.6
-
[4]
Zhao K J, Wang W L, Gregoire J, et al. Nano Lett., 2011, 11(7):2962-2967 doi: 10.1021/nl201501s
-
[5]
Verma P, Maire P, Novák P. Electrochim. Acta, 2010, 55(22):6332-6341 doi: 10.1016/j.electacta.2010.05.072
-
[6]
Liu B, Wang X F, Chen H T, et al. Sci. Rep., 2013, 3(15):1622-1628
-
[7]
Wu H, Chan G, Choi J W, et al. Nat. Nanotechnol., 2012, 7(5):310-315 doi: 10.1038/nnano.2012.35
-
[8]
Favors Z, Wang W, Bay H H, et al. Sci. Rep., 2014, 4(8):4605-4611
-
[9]
Deng J W, Ji H X, Yan C L, et al. Angew. Chem. Int. Ed., 2013, 125(8):2382-2386 doi: 10.1002/ange.201208357
-
[10]
Zhang T, Fu L J, Takeuchi H, et al. J. Power Sources, 2006, 159(1):349-352
-
[11]
Zhang T, Zhang H P, Yang L C, et al. Electrochim. Acta, 2008, 53(18):5660-5664 doi: 10.1016/j.electacta.2008.03.017
-
[12]
Li X L, Gu M, Hu S Y, et al. Nat. Commun., 2014, 5(5):4105-4111
-
[13]
Liu N, Lu Z D, Zhao J, et al. Nat. Nanotechnol., 2014, 9(3):187-192
-
[14]
Yao Y, Mcdowell M T, Ryu I, et al. Nano Lett., 2011, 11(7):2949-2954 doi: 10.1021/nl201470j
-
[15]
张兴帅, 许笑目, 郭玉忠, 等.无机化学学报, 2017, 33(3):377-382 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20170302ZHANG Xing-Shuai, XU Xiao-Mu, GUO Yu-Zhong, et al. Chinese J. Inorg. Chem., 2017, 33(3):377-382 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?file_no=20170302
-
[16]
Tang Y P, Yuan S, Guo Y Z, et al. Electrochim. Acta, 2016, 200:182-188 doi: 10.1016/j.electacta.2016.03.085
-
[17]
Gohier A, LaiK B, Kim K H, et al. Adv. Mater., 2012, 24(19):2592-2597 doi: 10.1002/adma.v24.19
-
[18]
Zhu B, Jin Y, Tan Y L, et al. Nano Lett., 2015, 15(9):5750-5754 doi: 10.1021/acs.nanolett.5b01698
-
[19]
Zhang T W, Hu L, Liang J W, et al. RSC Adv., 2016, 6(36):30577-30581 doi: 10.1039/C6RA00182C
-
[20]
Rey M, Elnathan R, Ditcovski R, et al. Nano Lett., 2015, 16(1):157-163
-
[21]
Zong L Q, Jin Y, Liu C, et al. Nano Lett., 2016, 16(11):7210-7215 doi: 10.1021/acs.nanolett.6b03567
-
[22]
Sandhage K H, Dickerson M B, Huseman P M, et al. Adv. Mater., 2002, 14(6):429-433 doi: 10.1002/(ISSN)1521-4095
-
[23]
Shi Y F, Zhang F, Hu Y S, et al. J. Cheminf., 2010, 132(16):5552-5553
-
[24]
Bao Z B, Weatherspoon M R, Shian S, et al. Nature, 2007, 38(23):172-175
-
[25]
Varanakkottu S N, Anyfantakis M, Morel M, et al. Nano Lett., 2016, 16(1):644-650 doi: 10.1021/acs.nanolett.5b04377
-
[26]
Ibisate M, Golmayo D, López C. Adv. Mater., 2009, 21(28):2899-2902 doi: 10.1002/adma.v21:28
-
[27]
Shepherd R F, Panda P, Bao Z H, et al. Adv. Mater., 2010, 20(24):4734-4739
-
[28]
Liu N, Huo K F, Mcdowell M T, et al. Sci. Rep., 2013, 3(5):1919-1925
-
[29]
Jung D S, Ryou M H, Sung Y J, et al. Proc. Natl. Acad. Sci. U.S.A., 2013, 110(30):12229-12234 doi: 10.1073/pnas.1305025110
-
[30]
Zhong Y, Shaw L L, Manjarres M, et al. J. Am. Ceram. Soc., 2010, 93(10):3159-3167 doi: 10.1111/jace.2010.93.issue-10
-
[31]
Zhang T W, Hu L, Liang J W, et al. RSC Adv., 2016, 6(36):30577-30581 doi: 10.1039/C6RA00182C
-
[32]
Favors Z, Wang W, Bay H H, et al. Sci. Rep., 2014, 4:5623-5629
-
[33]
Ahn J, Lee D H, Kang M S, et al. Electrochim. Acta, 2017, 245:893-901 doi: 10.1016/j.electacta.2017.05.164
-
[34]
Liang J W, Li X N, Hou Z G, et al. ACS Nano, 2016, 10(2):2295-2304 doi: 10.1021/acsnano.5b06995
-
[35]
Shi L, Wang W K, Wang A B, et al. J. Alloys Compd., 2016, 661:27-37 doi: 10.1016/j.jallcom.2015.11.196
-
[36]
Yoo J K, Kim J, Choi M J, et al. Adv. Energy Mater., 2015, 4(16):1400622
-
[37]
Su L W, Xie J, Xu Y W, et al. J. Alloys Compd., 2016, 663:524-530 doi: 10.1016/j.jallcom.2015.12.134
-
[38]
Liu Z L, Chang X H, Wang T, et al. ACS Nano, 2017, 11(6):6065-6073 doi: 10.1021/acsnano.7b02021
-
[39]
Florke O W, Graetsch H A, Brunk F, et al. Ullmanns Ency-clopedia of Industrial Chemicals. Hoboken, NJ:John Wiley and Sons, Inc., 2008:478-485
-
[40]
Park E, Yoo H, Lee J, et al. ACS Nano, 2015, 9(7):7690-7696 doi: 10.1021/acsnano.5b03166
-
[41]
Nguyen C C, Choi H, Song S W. J. Electrochem. Soc., 2013, 160(6):A906A914
-
[42]
Yasuda K, Kashitani Y, Kizaki S, et al. J. Power Sources, 2016, 329:462-472 doi: 10.1016/j.jpowsour.2016.08.110
-
[43]
许笑目, 张兴帅, 陈志柠, 等.高等学校化学学报, 2017, 38(5):713-721XU Xiao-Mu, ZHANG Xing-Shuai, CHEN Zhi-Ning, et al. Chem. J. Chinese Universities, 2017, 38(5):713-721

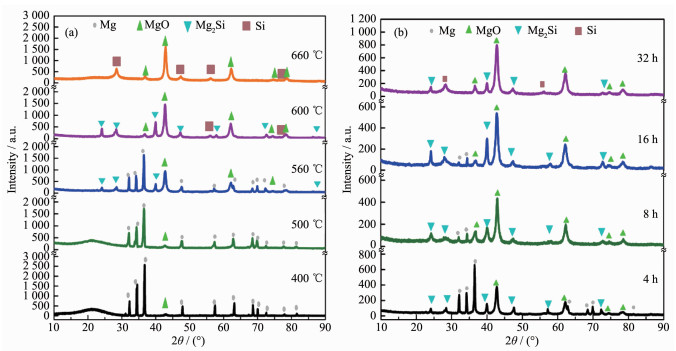
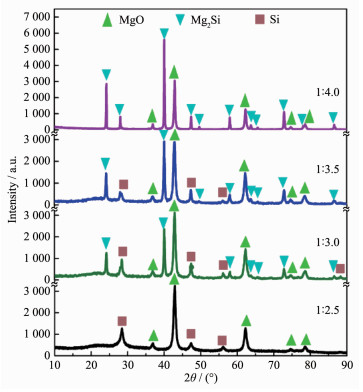
 下载:
下载:

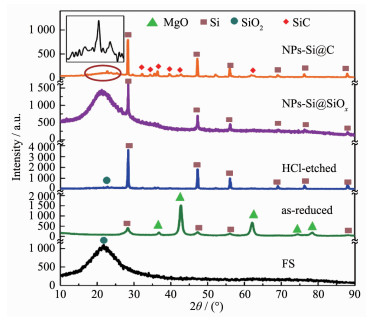


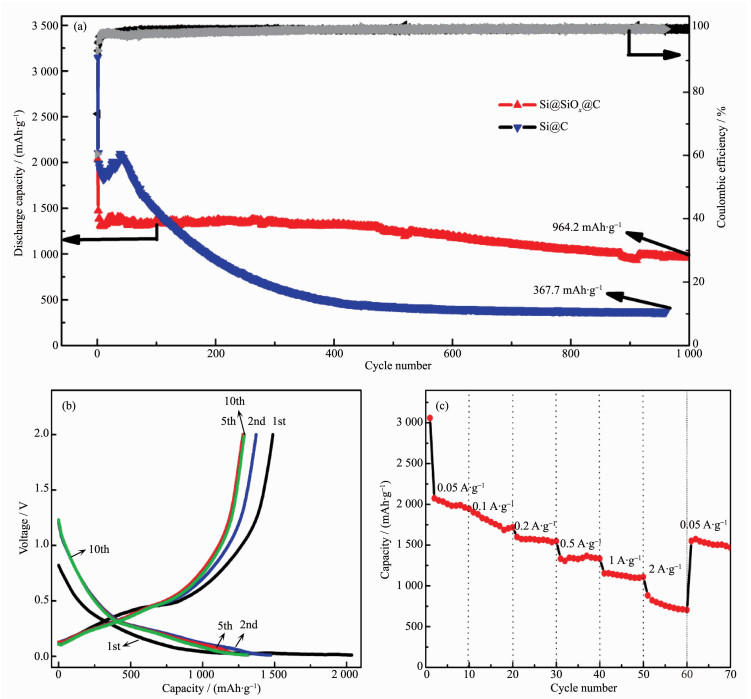

 下载:
下载:








 下载:
下载:
