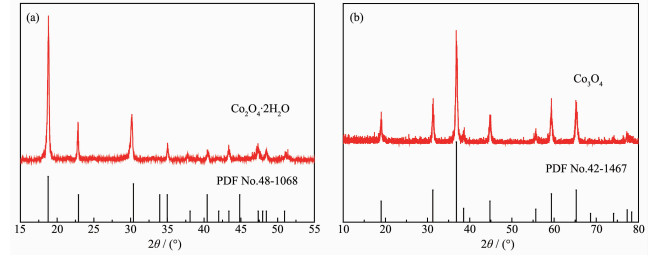
Figure 1.
PXRD patterns of CoC2O4·H2O precursors (a) and Co3O4 catalysts (b)
Catalytic Combustion of Ventilation Air Methane over Co3O4 Rectangular Prism
Ru-Yue NIU , Peng-Cheng LIU , Wei LI , Shuang WANG , Jin-Ping LI

As is known, methane is difficult to be oxidized due to the strongest C-H bond among hydrocarbons, however, it has been studied extensively over the past decades, and the catalytic combustion is still a challenging technology that can convert methane into carbon dioxide and water at relatively low temperature[1-4]. Especially, ventilation air methane (VAM) is much more difficult to be oxidized for its low concentration (0.1%~1.0% (V/V)) and fast flow rate[5]. Up to now, about 60%~70% methane is emitted to the atmosphere through VAM all over the world, particularly, in China, about 85%~90% of the total coal mine methane emissions mainly comes from VAM each year[6-7]. Moreover, the warming potential of methane is higher than that of carbon dioxide. Therefore, the efforts to remove methane from VAM and limit the emission of toxic gases become rather important[8].
Accordingly, catalysts for methane combustion undergo very demanding conditions: they must resist thermal, mechanical shocks and exhibit high activity. Noble metal-based catalysts, such as supported Pd and Pt catalysts, are well-known to be high activity at low temperature. Unfortunately, they are limited in industrial applications due to their high cost and low thermal stability[9-11]. Alternatively, oxide catalysts such as perovskites, hexaaluminates and transition metal oxides catalysts are the promising combustion catalysts with a low cost and relatively high thermal stability in methane combustion[12-15]. Compared to other oxide catalysts, cobalt oxide (Co3O4) is regarded as the most efficient catalyst, and many researchers identified that the activity for methane combustion follows the order: Co3O4 > CuO >NiO > Fe2O3 > Mn2O3 > Cr2O3[16-18].
Generally, the morphologies and the crystal planes of Co3O4 nanocrystals have promoting performances in methane combustion. Chen et al.[19] reported that the Co3O4 catalysts containing {111} planes exhibit the higher catalytic activity than the {100} planes for the methane combustion, which confirms the effect of crystal planes on the catalytic performance. Moreover, some researches show that the high Co3+/Co2+ ratio is favorable to the methane combustion, because it can facilitate desorption of oxygen and in turn assist methane combustion[20-21]. Furthermore, the adsorbed oxygen species play an important role in catalytic oxidation reactions[22-24]. For example, Fei et al.[25] suggested that the Co3O4 nanotubes have higher catalytic activity than the Co3O4 nanoparticles in the methane combustion, because the nanotubes Co3O4 possess much more adsorbed oxygen.
In the current work, a Co3O4 rectangular prism catalyst was prepared by a two-step method, and its catalytic activity was evaluated by the temperature of methane combustion in VAM. The relationship between the structure and catalytic performance of the above-mentioned catalyst is well studied based on plenty of characterizations such as PXRD, SEM, HRTEM, N2 adsorption-desorption, H2-TPR, and XPS.
All the reagents were obtained from commercial sources and used without further purification. Cobalt(Ⅱ) acetate tetrahydrate (99.5%) was purchased from Sinopharm Chemical Reagent Co., Ltd. Oxalic acid (99.0%) was purchased from Tianjin Chemical Reagent Co., Ltd. Hexamethylenetetramine (99.0%) was purchased from Tianjin Beichen Founder Reagent Factory. All used gases were high pure gases (99.99%).
In a typical synthesis procedure, 20 mmol of Cobalt(Ⅱ) acetate tetrahydrate was dissolved in 100 mL of distilled water at 40 ℃. Then 10 mL of an aqueous solution containing 20 mmol of hexamethylenetetramine and 4 mmol of oxalic acid was added dropwise under stirring within two minutes. The pink turbidity solution was refluxed at 95 ℃ for 6 h and a light pink Co-based precursor was obtained. The Co3O4 was made by heating the precursor at 350 ℃ in air for 2 h.
The phase purity and crystal structure of the catalysts were examined on a Rigaku Mini Flex Ⅱ benchtop X-ray diffractometer using Cu Kα radiation (30 kV, 15 mA, λ=0.154 18 nm) in the 2θ range of 10°~80° with a step size of 0.01° and a scanning rate of 8°·min-1.
Morphologies of the samples were observed by SEM (Hitachi, SU8010, 3 kV). The HRTEM measurement was carried out with FEI Tecnai G2 F20 S-Twin equipment operated at an accelerating voltage of 200 kV. The catalyst powder was ultrasonically dispersed in ethanol and dropped onto a copper grid coated with amorphous carbon film, then dried in air.
The Brunauer-Emmett-Teller (BET) surface area and pore size distribution of the catalyst were measured with a Micromeritics TriStar Ⅱ 3020 instrument using adsorption of N2 at 77 K. Before each adsorption experiment, the catalyst was heated at 200 ℃ under vacuum for 3 h. Barrett-Joyner-Halenda (BJH) method was used to calculate the pore size distribution from desorption branch of the isotherm.
The X-ray photoelectron spectroscopy (XPS) test was performed on an ESCALAB 220i-XL spectrometer by using Al Kα (1 486.6 eV) as the X-ray source. The equipment base pressure was 3×10-5 Pa, and the sample was characterized at room temperature. Detailed spectra were recorded for the region of Co2p and O1s photo-electrons with a 0.1 eV step. Analysis was performed by the XPS Peak Fit software, and charging effects were corrected by adjusting binding energy (B.E.) of C1s (284.6 eV).
The H2-temperature programmed reduction (H2-TPR) was analyzed with a Micromeritics AutoChem Ⅱ 2920 instrument. Prior to H2-TPR experiment, 50 mg catalyst was purged in flowing Ar at 200 ℃ for 1 h with a total flow rate of 30 mL·min-1, then cooled down to 50 ℃ in Ar flow. The reduction process was carried out in the temperature range of 50~900 ℃ in H2/Ar (VH2/VAr=10%, 30 mL·min-1). The hydrogen consumption was estimated from the area under the peak after taking the thermal conductivity detector response into consideration. Calibration of thermal conductivity detector (TCD) signal has been done with an Ag2O standard (Merck, reagent grade). The data processing has been done by using Origin Pro 8.0 program, which allows the deconvolution of the temperature-programmed reduction (TPR) peaks in well-defined Gaussian-shaped components.
The methane combustion on Co3O4 rectangular prism catalyst was carried out at atmospheric pressure in a conventional flow system using a fixed-bed quartz micro-reactor (length=400 mm, inner diameter=6 mm). A gas mixture consisted of CH4, O2 and N2 (VCH4:VO2:VN2=1:20:79) was introduced into the quartz micro-reactor at a total flow rate of 40 mL·g-1·h-1 corresponding to a gas hourly space velocity (GHSV) of 16 000 mL·g-1·h-1. When GHSV changed from 16 000 to 112 500 mL·g-1·h-1, the total flow rate was varied from 40 to 150 mL·g-1 ·h-1. According to different GHSVs, 80~150 mg of catalysts (20~40 mesh) were loaded in the quartz tube micro-reactor, respectively. Prior to each measurement, the catalyst was pretreated at 200 ℃ for 1 h with a nitrogen flow of 30 mL·min-1. Activity data were obtained at steady state condition from 200 to 450 ℃ while increasing the temperature by 50 ℃.The effluent gases were analyzed online with a gas chromatograph (ZHONGKEHUIFEN GC-6890A) equipped with a TDX-01 column and a thermal conductivity detector. The methane conversion (XCH4) was calculated according to the following equation: XCH4=(X0-XT)/X0×100%, where X0 refers to the volumetric concentration of methane in the feed and XT corresponds to the concentration of methane at the given temperature. In all tests, CO2 and H2O were the only detected products in the exhaust stream during reaction, and CO was not found in the effluent gases, implying the conversion of methane to carbon dioxide. For comparison, commercial Co3O4 was also investigated.
Fig. 1a presents the PXRD patterns of as-prepared CoC2O4·2H2O precursors. The presence of peaks at 2θ=18.7°, 22.7°, 30.1°, 35.0°, 37.6°, 40.4°, 43.3°, 47.3°, 48.4°, and 51.1° could be assigned to the (202), (004), (400), (022), (206), (315), (224), (602), (026) and (130) planes of CoC2O4·2H2O (PDF No.48-1068). In the Fig. 1b, the diffraction peaks of Co3O4 rectangular prism at 19.0°, 31.2°, 36.6°, 38.5°, 44.8°, 55.7°, 59.4° and 65.3° could be assigned to the (111), (220), (311), (222), (400), (422), (511) and (440) planes of the spinel phase Co3O4 (PDF No.42-1467). In the case of the Co3O4 sample, no other peaks can be detected for impurities, which indicate that the sample consists of pure Co3O4 phase.
In order to have a better understanding of the morphological and structural, a detailed microscopy investigation by SEM and high-resolution analysis are performed on both CoC2O4·2H2O precursors and Co3O4 catalysts. Fig. 2 displays SEM imags of the as-prepared CoC2O4·2H2O at 95 ℃ and the corresponding Co3O4 products after calcination. As shown in Fig. 2a, most CoC2O4·2H2O are uniform rectangular prisms with smooth surfaces, and the size distribution is in the range of 2~5 μm (Fig. 2b). Fig. 2(c, d) show the SEM images of the prepared Co3O4 catalysts after calcination at 350 ℃. It is found that most of the Co3O4 catalysts well maintain the rectangular prism shape. Fig. 3 shows the HRTEM images of Co3O4 catalysts. Seen in Fig. 3a, the Co3O4 catalyst is formed by the accumulation of small particles of 10~20 nm, ultrasonication results in the breakdown of rectan-gular prisms into nanoparticles. Their lattice fringes are clear (Fig. 3b), which are attributed to (220) planes with a lattice space of 0.278 nm. The dominant exposed plane of Co3O4 rectangular prisms is {111} planes, which is the plane normal to the set of (220) planes.
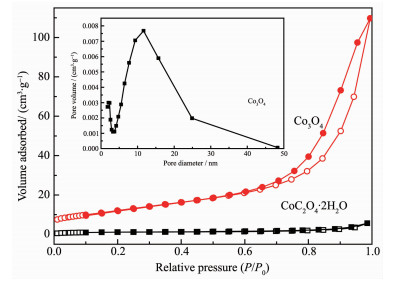
To further investigate the specific surface areas and the porous nature of the CoC2O4·2H2O precursors and Co3O4 catalysts, Nitrogen adsorption-desorption isotherms of the two samples are shown in Fig. 4, and the insets illustrate the corresponding Barrett-Joyner-Halenda (BJH) pore size distribution plots. For Co3O4 catalysts, nitrogen adsorption experiment has given a typical type-Ⅳ isotherm with a distinct hysteresis loop observed in the relative pressure (P/P0) range of 0.7~1.0, which is the characteristic of mesoporous materials. The BET surface area for the CoC2O4·2H2O precursors and Co3O4 catalysts are found to be about 4 and 45 m2·g-1, respectively. The increase of BET surface area may result from the decomposition of the CoC2O4·2H2O precursors. Moreover, the BET surface area of the commercial Co3O4 catalyst is measured to be 1 m2·g-1. According to the BJH plot calculated from the nitrogen isotherm, the average pore diameter of Co3O4 catalysts is about 14 nm, which indicated that the sample contains mesoscale pores.
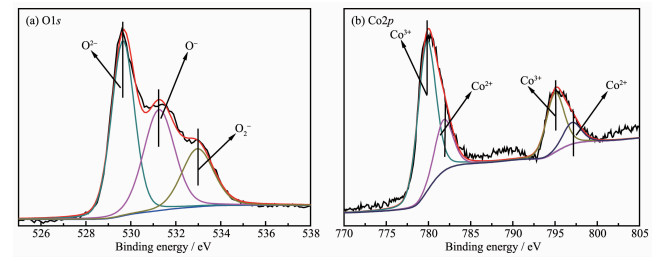
XPS analysis was performed in order to gain the binding energy and the percentages of surface atoms. O1s and Co2p photoelectron spectra for the rectangular prism Co3O4 are shown in Fig. 5. As indicated in Fig. 5a, three peaks have been observed. The peak at ~529.7 eV (O2-) is attributed to lattice oxygen species (Olat) of the catalyst. And the peaks at ~531.3 (O-) and ~533.0 eV (O2-) are attributed to the adsorption oxygen species (Oads)[26]. According to literatures, higher relative concentration ratio of Olat/Oads is previously found to be preferable for methane combustion. The ratio of the peak intensities of the surface-adsorbed oxygen species to lattice oxygen is 1.24, thus high activity for methane combustion can be obtained[27-28].
Two sharp peaks at 795.2 and 780.0 eV correspond to the Co2p1/2 and Co2p3/2 spin-orbit-split doublet peak of Co3O4 spinel, respectively. There is an energy difference of approximately 15.2 eV between them. The same chemical information can be obtained by analyzing the Co2p1/2 and Co2p3/2 spectra. Therefore, only Co2p3/2 peaks in Fig. 5b are fitted and de-convoluted into two peaks at 781.8 and 779.8 eV, which are attributed to Co2+ and Co3+ [28-29], respectively. Co3O4, containing a Co3+/Co2+ couple, is favorable to methane combustion. Moreover, the main oxidation state of Co in the Co3O4 rectangular prism is Co3+ (the ratio of the peak intensities of Co3+ to Co2+ is 2.87), accordingly, higher oxidation state of Co species was previously found to be preferable for oxidation reactions over the Co containing catalysts[19, 30].
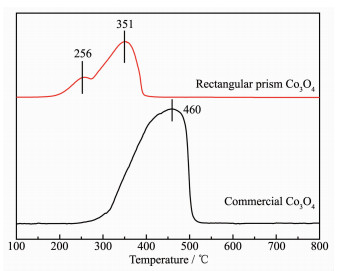
To investigate the reducibility of the Co species in Co3O4 rectangular prism and commercial Co3O4 catalysts, H2-TPR experiments are carried out. The reduction profiles of the samples have been displayed in Fig. 6. In this case, the Co3O4 rectangular prism contains two reduction peaks, the first peak is at 256 ℃, which is associated with the reduction of Co3O4 to CoO, and the second broad peak at 351 ℃ is correspond to the reduction of CoO to Co. However, the commercial Co3O4 catalysts have a wide reduction peak centered at 460 ℃. The results show that the performance of Co3O4 rectangular prism is better than that of commercial Co3O4 catalysts. Accordingly, the same oxide species, which has the lower reduction temperature, owing the easier activation of bond metal-oxygen (Co-O)[31-32].
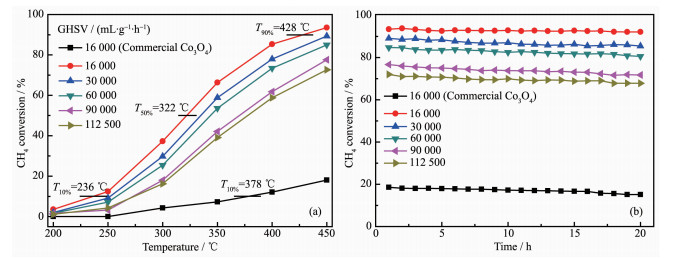
The activity in methane complete oxidation was expressed in terms of methane conversion with respect to the reaction temperature for Co3O4. Accordingly, the catalytic activity of methane oxidation is evaluated by the light-off temperature (T10%), the half-conversion temperature (T50%) and the total conversion temperature (T90%), representing the temperature of methane conversions at 10%, 50% and 90%, respectively. The T10%, T50% and T90% over the rectangular prism Co3O4 catalyst are 236, 322 and 428 ℃ at 16 000 mL·g-1·h-1, respectively (Fig. 7a). However, the T10% over commercial Co3O4 catalyst is 378 ℃, in addition, the highest methane conversion merely reached to 18.06% at 450 ℃. Moreover, the methane conversion over the rectangular prism Co3O4 catalyst increased with increasing temperature from 200 ~450 ℃. To convert low-concentration methane effectively by catalytic oxidation in practical application, GHSV is a critical parameter. Thus, the effects of GHSVs on methane conversion over the Co3O4 rectangular prism catalyst were studied. A general change is observed from Fig. 7a, that is, methane conversion rates decrease as increasing GHSVs. When the GHSVs are 30 000, 60 000, 90 000 and 112 500 mL·g-1·h-1, the methane conversions are decreased 4.16%, 8.54%, 15.89% and 20.85% compared to 16 000 mL·g-1·h-1 at 450 ℃, respectively. In general, a high GHSV has provided a short residence time and frequent contacts between catalyst and reaction gases, which leads to the decrease in the methane conversions. This result displays that enough contact time is necessary for enhancing the catalytic activity. As seen in Fig. 7b, the catalytic stability of the rectangular prism Co3O4 catalyst was examined at different GHSVs. After the samples were operated at 450 ℃ for 20 h under different GHSVs, the methane conversion rates only have slight decreases. For comparison, when the GHSV was 16 000 mL·g-1·h-1, the catalytic stability of the commercial Co3O4 catalyst was tested. It can be seen that the methane conversion rates keep at approximately 16% within running stable for 20 h.
In summary, a novel Co3O4 rectangular prism with excellent activity and good stability towards the methane combustion has been synthesized through a two-steps method. The superior catalytic activity can be attributed to the following reasons: firstly, the Co3O4 rectangular prism dominantly exposed the {111} crystal planes, which confirmed the effect of crystal planes on the methane combustion performance; Secondly, high surface Co3+ content and high content surface adsorbed oxygen both play crucial roles in the methane catalytic oxidation. Owing to its simplicity of synthesis, low cost and excellent methane combustion performance, the novel Co3O4 rectangular prism could be a very important and promising heterogeneous catalyst.
O'malley A, Hodnett B K. Catal. Today, 1999, 54(1):31-38 doi: 10.1016/S0920-5861(99)00166-2
任晓光, 葛秀涛.无机化学学报, 2012, 28(4):823-828 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20120428&journal_id=wjhxxbcnREN Xiao-Guang, GE Xiu-Tao. Chinese J. Inorg. Chem., 2012, 28(4):823-828 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20120428&journal_id=wjhxxbcn
Kirchnerova J, Alifanti M, Delmon B. Appl. Catal., A, 2002, 231(1/2):65-80 http://www.sciencedirect.com/science/article/pii/S0926860X01009036
陈永东, 朱艺, 王健礼, 等.无机化学学报, 2009, 25(10):1771-1778 doi: 10.3321/j.issn:1001-4861.2009.10.012CHEN Yong-Dong, ZHU Yi, WANG Jian-Li, et al. Chinese J. Inorg. Chem., 2009, 25(10):1771-1778 doi: 10.3321/j.issn:1001-4861.2009.10.012
Guo T Y, Du J P, Wu J T, et al. Chem. Eng. J., 2016, 306:745-753 doi: 10.1016/j.cej.2016.08.020
Ouyang S, Xu S, Song N, et al. Fuel, 2013, 113:420-425 doi: 10.1016/j.fuel.2013.06.004
Cheng Y P, Wang L, Zhang X L. Int. J. Greenhouse Gas Control, 2011, 5(1):157-166 doi: 10.1016/j.ijggc.2010.07.007
Guo T Y, Du J P, Li J P. J. Mater. Sci., 2016, 51(24):10917-10925 doi: 10.1007/s10853-016-0303-z
Lovón-Quintana J J, Santos J B O, Lovón A S P, et al. J. Mol. Catal. A:Chem., 2016, 411:117-127 doi: 10.1016/j.molcata.2015.08.001
Ruiz J A C, Oliveira E C, Fraga M A, et al. Catal. Commun., 2012, 25:1-6 doi: 10.1016/j.catcom.2012.04.006
Chen C, Yeh Y H, Cargnello M, et al. ACS Catal., 2014, 4(11):3902-3909 doi: 10.1021/cs501146u
Cao X X, Zhou R, Rui N, et al. Catal. Today, 2017, 297:219-227 doi: 10.1016/j.cattod.2017.01.042
缪建文, 范以宁, 金永漱, 等.无机化学学报, 2003, 19(12):1361-1365 doi: 10.3321/j.issn:1001-4861.2003.12.019MIU Jian-Wen, FAN Yi-Ning, JIN Yong-Shu, et al. Chinese J. Inorg. Chem., 2003, 19(12):1361-1365 doi: 10.3321/j.issn:1001-4861.2003.12.019
Hu R S, Ding R R, Chen J, et al. Catal. Commun., 2012, 21:38-41 doi: 10.1016/j.catcom.2012.01.008
郑建东, 葛秀涛.无机化学学报, 2013, 29(6):1307-1311 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20130631&journal_id=wjhxxbcnZHENG Jian-Dong, GE Xiu-Tao. Chinese J. Inorg. Chem., 2013, 29(6):1307-1311 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20130631&journal_id=wjhxxbcn
Xu X L, Han H, Liu J J, et al. J. Rare Earths, 2014, 32(2):159-169 doi: 10.1016/S1002-0721(14)60046-6
Zavyalova U, Scholz P, Ondruschka B. Appl. Catal., A, 2007, 323:226-233 doi: 10.1016/j.apcata.2007.02.021
Hu L H, Peng Q, Li Y D. J. Am. Chem. Soc., 2008, 130(48):16136-16137 doi: 10.1021/ja806400e
Chen Z P, Wang S, Liu W G, et al. Appl. Catal., A, 2016, 525:94-102 doi: 10.1016/j.apcata.2016.07.009
Zou G C, Chen M X, Shangguan W F. Catal. Commun., 2014, 51:68-71 doi: 10.1016/j.catcom.2014.03.028
Wang Q, Peng Y, Fu J, et al. Appl. Catal., B, 2015, 168:42-50
Chen Z P, Wang S, Ding Y, et al. Appl. Catal., A, 2017, 532:95-104 doi: 10.1016/j.apcata.2016.12.021
Wang F G, Zhang L J, Xu L L, et al. Fuel, 2017, 203:419-429 doi: 10.1016/j.fuel.2017.04.140
Huang H, Dai Q G, Wang X Y. Appl. Catal., B, 2014, 158:96-105 http://www.sciencedirect.com/science/article/pii/S0926337314000824
Fei Z Y, He S C, Li L, et al. Chem. Commun., 2012, 48(6):853-855 doi: 10.1039/C1CC15976C
Yang N T, Ni S L, Sun Y H, et al. Mol. Catal., 2018, 452:28-35 doi: 10.1016/j.mcat.2018.03.016
Petitto S C, Marsh E M, Carson G A, et al. J. Mol. Catal., A, 2008, 281(1/2):49-58 http://www.sciencedirect.com/science/article/pii/S1381116907005067
Wei Y C, Ren X, Ma H M, et al. Chem. Commun., 2018, 54(12):1533-1536 doi: 10.1039/C7CC08423D
Yang H, Yang W H, Lv K L, et al. Microporous Mesoporous Mater., 2018, 255:36-43 doi: 10.1016/j.micromeso.2017.07.016
Li Y, Shen W J. Chem. Soc. Rev., 2014, 43(5):1543-1574 doi: 10.1039/C3CS60296F
Zhu Z Z, Lu G Z, Zhang Z G, et al. ACS Catal., 2013, 3(6):1154-1164 doi: 10.1021/cs400068v
Yan Q Y, Li X Y, Zhao Q D, et al. J. Hazard. Mater., 2012, 209:385-391 http://www.sciencedirect.com/science/article/pii/S0304389412000702

Figure 4 N2 adsorption-desorption isotherms of Co3O4 catalysts and CoC2O4·H2O precursors, and pore diameter distribution (inset) of Co3O4

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载:



 下载:
下载:





 下载:
下载: