表 1
Crystal data and structure refinement for 1
Table 1.
Crystal data and structure refinement for 1
Synthesis, Crystal Structure and Magnetism of a[Dy2Co8] Core Complex with Diethanolamine Ligand
You-Zhu YU , Yu-Hua GUO , Yan-Hong SHEN , Li-Guo YANG , Yong-Sheng NIU , Xiao-Ming ZHENG , Hai-Hui ZHANG , Fang WANG

The study of 3d-4f heterometallic complexes is an active area of research in contemporary coordination chemistry because of their potential applications in various fields such as non-linear optical materials, luminescence, molecular adsorption and magnetism[1-4]. Importantly, some of these 3d-4f heterometallic complexes behave single-molecule magnets (SMMs) properties. Most notable in this vein is the research on polynuclear metal complexes, many of which display fascinating molecular structures and, more importantly, interesting magnetic properties due to the unique exchange interactions between the metal ions[5-10]. Over recent years, many examples with SMM behavior of 3d-4f compounds involving cobalt(Ⅱ) ion have been reported[11-15]. It is meaningful to continue to construct and study the magnetism of Co-Ln based molecular cluster. Using bridging ligands to bind 3d and 4f paramagnetic building blocks is the main synthetic plan to develop discrete polynuclear complexes and improve magnetic properties for 3d-4f complexes. Diethanolamine was usefully selected to construct molecular clusters because of its intrinsic nature, such as having more coordination sites and good flexibility[16-18]. In this paper, we report the syn-thesis, structure and preliminary magnetic studies for a new type of [Dy2Co8] complex [Dy2Co8(L)4(HL)4(HCOO)4(OH)2Cl2(CH3OH)2]Cl2·4CH3OH·2H2O using diethanolamine as the ligand.
All starting reagents were of A.R. grade and used as purchased without further purification. IR spectrum was recorded as KBr discs on a Shimadzu IR-408 infrared spectrophotometer in the 4 000~600 cm-1 range. Analyses of C, H and N were determined on a Perkin-Elmer 240 Elemental analyzer. Thermogravi-metric analysis (TGA) experiments were carried out on a NETZSCH STA 449F3 thermal analyzer from 40 to 800 ℃ under N2 at a heating rate of 10 ℃·min-1. Powder X-ray diffraction (PXRD) determinations was performed on an X-ray diffractometer (D/max 2500 PC, Rigaku) with Cu Kα radiation (λ=0.154 06 nm). The crushed single crystalline powder samples were scanned at 40 kV (generator voltage) and 40 mA (tube current) from 5° to 50° with a step of 0.1°·s-1. The magnetic susceptibility measurements was measured over the temperature range of 2~300 K with a Quantum Design MPMS-XL7 SQUID magnetometer using an applied magnetic field of 2 000 Oe.
To a stirred solution of DyCl3·6H2O (0.3 mmol, 113 mg) and CoCl2·6H2O (1 mmol, 238 mg) in CH3OH (15 mL) was added diethanolamine (2 mmol, 210 mg). After 15 min, CH3ONa (1 mmol, 54 mg) were added to the above solution, and the resulting mixture was stirred for 6 h to give a dark-red solution that was filtered. The dark-red filtrate was then left undi-sturbed for 1 week. Dark-red, cube-shaped crystals were retrieved in 40% yield (based on Dy). Anal. Calcd. for C42H110Cl4Co8Dy2N8O34(%): C, 22.83; H, 5.02; N, 5.07. Found(%): C, 22.87; H, 5.01; N, 5.14.
Single-crystal X-ray diffraction data of complex 1 was collected on a Bruker SMART APEX Ⅱ diffra-ctometer equipped with a graphite-monochromatized Mo Kα radiation (λ=0.071 073 nm) at room tempera-ture by using an ω-2θ scan mode. The structure was solved by directmethods with SHELXS-97 program[19] and refined by full-matrix least-squares techniques on F2 with SHELXL-97[20]. All non-hydrogen atoms were refined anisotropically. H atoms attached to C were placed geometrically and allowed to ride during subsequent refinement with an isotropic displacement parameter fixed at 1.2 times Ueq of the parent atoms. H atoms bonded to N or O atoms were first located in difference Fourier maps and then placed in the calculated sites and included in the refinement. The crystallographic data and selected bond lengths and angles are listed in Table 1 and Table 2, respectively.
 下载:
导出CSV
下载:
导出CSV
| Empirical formula | C42H110Cl4Co8Dy2N8O34 |
| Formula weight | 2 201.55 |
| Crystal size/mm | 0.15×0.14×0.12 |
| Temperature/K | 296(2) |
| Crystal system | Triclinic |
| space group | P1 |
| a/nm | 1.160 4(8) |
| b/nm | 1.322 7(9) |
| c/nm | 1.465 3(9) |
| α/(°) | 72.181(7) |
| β/(°) | 67.638(7) |
| γ/(°) | 65.939(7) |
| V/nm3 | 1.869(2) |
| Z | 1 |
| Dc/(g·cm-3) | 1.956 |
| μ(Mo Kα)/mm-1 | 3.928 |
| F(000) | 1 098 |
| θ range/(°) | 2.491~25.997 |
| Reflection collected | 28 782 |
| Independent reflection (Rint) | 7 318 (0.051 1) |
| Data, restrain, parameter | 7 318, 38, 442 |
| Limiting indices (h, k, l) | -14~14, -16~16, -18~18 |
| R1, wR2 [I > 2σ(I)] | 0.056 1, 0.153 1 |
| R1, wR2 (all data) | 0.077 7, 0.199 6 |
| S on F2 | 1.147 |
| Largest diff. peak and hole/(e·nm-3) | 3 700 and -2 392 |
下载:
导出CSV
| Dy(1)-O(9) | 0.229 7(5) | Co(3)-O(4) | 0.188 0(6) | Co(2)-O(10) | 0.194 1(6) |
| Dy(1)-O(12) | 0.237 8(6) | Co(3)-N(2) | 0.192 8(9) | Co(2)-N(3) | 0.196 3(8) |
| Dy(1)-O(7) | 0.240 3(8) | Co(1)-O(9) | 0.205 8(6) | Co(4)-O(2) | 0.206 0(6) |
| Dy(1)-O(4) | 0.240 4(6) | Co(1)-O(8) | 0.208 8(7) | Co(4)-Cl(1) | 0.236 9(4) |
| O(9)-Dy(1)-O(12) | 125.6(2) | O(9)-Co(1)-O(8) | 94.9(3) | N(2)-Co(3)-N(1) | 100.0(4) |
| O(9)-Dy(1)-O(7) | 76.4(2) | O(12)-Co(2)-O(11) | 177.3(3) | O(2)-Co(4)-O(6) | 150.0(3) |
| O(12)-Dy(1)-O(7) | 119.5(3) | O(4)-Co(3)-O(2) | 175.5(3) | ||
| O(7)-Dy(1)-O(4) | 72.6(2) | O(4)-Co(3)-N(2) | 86.6(3) |
CCDC: 1576526.
The FT-IR spectra of 1 (in KBr) show the bands as follows: 3 400(s), 3 161(s), 2 935(w), 2 870(w), 1 598(s), 1 346(s) and 1 042(s) cm-1. The absorption peak between 1 690 and 1 730 cm-1 is not observed, showing all carboxylic groups are deprotonated[21]. The strong peaks at 1 598 and 1 346 cm-1 are the νas(COO-) and νs(COO-) stretching mode of the coordinated HCOO- ligand. Peak at 1 042 cm-1 could be attributed to the stretching vibration of C-O bond. In addition, the strong and broad band centered at 3 400 cm-1 for the title compound is attributable to the O-H or N-H stretching vibration on the basis of known structure.
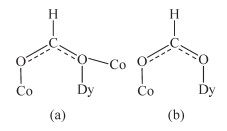
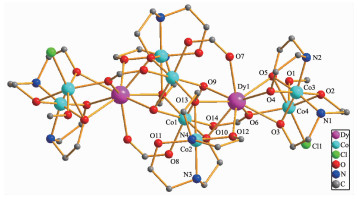
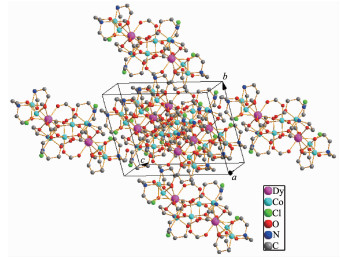
The crystal structure of 1 is revealed in Fig. 1, and its stacking X-ray structure in Fig. 2. The title compound, crystallized as [Dy2Co8(L)4(HL)4(HCOO)4(OH)2Cl2(CH3OH)2]Cl2·4CH3OH·2H2O, belongs to the triclinic system and P1 space group. The structure of this complex is centrosymmetric and contains two Dy(Ⅲ) and eight Co(Ⅱ) ions. Each Dy(Ⅲ) was nine-coordinated by nine oxygen atoms with the average Dy-O bond length 0.244 5 nm. Each Co(Ⅱ) ion displays distorted octahedral coordination environment. The ligand diethanolamine plays very important role in construction the structure via bridging and chelating metal atoms, and the nitrogen atoms only participate the coordination of cobalt atoms. Four coordination modes were found in the structure. In this case, a, b and c present μ3-η1: η2: η3 coordination mode, while d presents μ3-η1: η1: η3 coordination mode (Scheme 1). In the structure two coordination modes of HCOO- were found which mostly come from the oxidation of the solvent methanol(Scheme 2). There are two same μ3-OH groups in the structure which bridges two cobalt atoms and one dysprosium atom. Two kinds of Cl- ions were found in the structure, one of which involved in coordination, and another is used to balance the charge. Also, there are four methanol and two water molecules contained in the stucture as the solvent molecules. The existence of some O-H and N-H bonds makes more hydrogen bonds in the structure (Table 3).

 下载:
导出CSV
下载:
导出CSV
| D-H…A | d(D-H)/nm | d(H…A)/nm | d(D…A)/nm | ∠DHA/(°) |
| O(1W)-H(1WA)…O(11)#1 | 0.096 | 0.209 | 0.270 5(11) | 120.3 |
| O(16)-H(16C)…O(1W)#2 | 0.096 | 0.192 | 0.272 8(17) | 140.2 |
| O(15)-H(15C)…N(3)#3 | 0.096 | 0.222 | 0.295 0(13) | 132.4 |
| C(7)-H(7B)…O(1W)#4 | 0.097 | 0.261 | 0.351 1(19) | 154.7 |
| C(21)-H(21A)…Cl(2)#2 | 0.096 | 0.269 | 0.360(2) | 157.3 |
| C(4)-H(4A)…Cl(1) | 0.097 | 0.292 | 0.359 6(12) | 127.3 |
| C(6)-H(6A)…O(1) | 0.097 | 0.26 | 0.319 3(15) | 119.8 |
| C(16)-H(16B)…O(4) | 0.097 | 0.253 | 0.313 8(12) | 120.5 |
| C(14)-H(14A)…Cl(2)#1 | 0.097 | 0.267 | 0.361 9(12) | 166.8 |
| C(9)-H(9A)…O(7) | 0.097 | 0.249 | 0.307 1(13) | 118.2 |
| C(3)-H(3B)…Cl(2)#4 | 0.097 | 0.291 | 0.381 7(14) | 156.8 |
| C(12)-H(12A)…O(14)#1 | 0.097 | 0.265 | 0.330 7(12) | 125.4 |
| O(1)_H(1C)…O(1W) | 0.085 | 0.196 | 0.262 1(11) | 133.4 |
| O(9)-H(9C)…O(11)#1 | 0.098 | 0.175 | 0.262 5(8) | 146.6 |
| Symmetry transformations used to generate equivalent atoms: #1: -x+1, -y+1, -z+1; #2: x+1, y, z; #3: x, y, z+1; #4: -x+1, -y, -z+1. | ||||
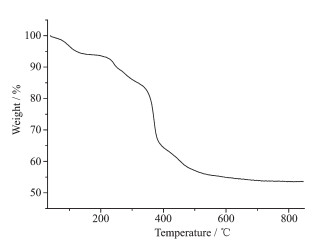
Thermal stability is an important aspect for the application of coordination compound. Thermogravi-metric analysis (TGA) experiments were carried out to determine the thermal stabilities of 1 (Fig. 3). TG curves showed the first consecutive step of weight loss was observed in the range of 50~180 ℃, corresponding to the release of solvent molecules (Calcd. 7.4%; Obsd. 6.9%). Then, the continuously weight loss corresponds to the release of ligands (in the range of 180~600 ℃). Finally, the weight loss ends until heating to 600 ℃ and the total weight loss was about 45%.
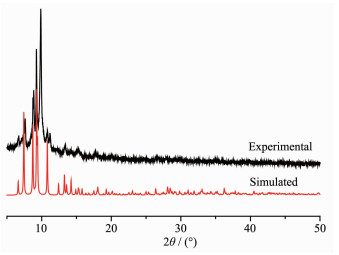
Powder X-ray diffraction (PXRD) experiment for complex 1 has also been conducted to confirm whether the crystal structures are truly consistent with the bulk materials. The PXRD computer-simulated and experimental patterns of complex 1 are shown in Fig. 4. In comparison with the simulated from crystal mode, the bulk-synthesized material and as-grown crystal can be considered homogeneous for complex 1.
Temperature-dependent direct current (DC) mag-netic susceptibility measurements for 1 was performed at temperatures ranging from 2 to 300 K, under an applied field of 1 000 Oe. The room temperature χmT value (40.81 cm3·mol-1·K) (Fig. 5) is slightly smaller than spin only value (43.34 cm3·mol-1·K) for two Dy(Ⅲ) (6H15/2, g=4/3) and eight Co(Ⅱ) (g=2, S=3/2), remaining constant before decreasing from 40.57 cm3·mol-1·K at 100 K to 38.47 cm3·mol-1·K at 15 K indi-cative of Stark level splitting in the lanthanide ions. Below 15 K, χmT value increases rapidly to 40.05 cm3·mol-1·K at 4.5 K before dropping to 37.09 cm3·mol-1·K at 2 K. The increase of magnetic susceptibility data at low temperature reveals the weak ferromag-netic interactions between metal centers or strong orbital contribution from Co(Ⅱ) ions.

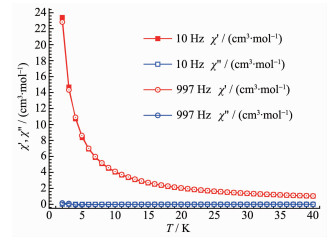
Field-dependent magnetization (M) dada for 1 was collected in the range of 0~5 T at 2~5 K for the title compound (Fig. 6). The curves of M vs H exhibit a gradual increase with increasing field strength reaching a saturation M value of 16.5μB at 2 K and 5 T, smaller than the theoretical saturated value for two Dy(Ⅲ) and eight Co(Ⅱ) ions. The magnetic anisotropy, together with the strong spin-orbital coupling, should be responsible for the magnetic unsaturation even at 5 T. This suggests the presence of magnetic anisotropy and/or considerable crystal-field effects. AC susceptibility measurement has also been conducted (Fig. 7). However, no in-phase and out-of-phase signal could be observed.
In summary, we have successfully prepared an unprecedented [Dy2Co8] core complex with diethanola-mine as ligand, which have been structurally chara-cterized. In particular, magnetic properties of the title complex was investigated and AC susceptibility measurement revealed that in-phase and out-of-phase signal could not be observed.
Sessoli R, Powell A K. Coord. Chem. Rev., 2009, 253(19):2328-2341 http://www.sciencedirect.com/science/article/pii/S0010854508002440
Andruh M. Chem. Commun., 2007(25):2565-2577 doi: 10.1039/b616972d
Sorace L, Benelli C, Gatteschi D. Chem. Soc. Rev., 2011, 40(6):3092-3104 doi: 10.1039/c0cs00185f
Chen W P, Liao P Q, Yu Y, et al. Angew. Chem., Int. Ed., 2016, 55(32):9375-9379 doi: 10.1002/anie.201603907
Kong X J, Ren Y P, Long L S, et al. J. Am. Chem. Soc., 2007, 129(22):7016-7017 doi: 10.1021/ja0726198
Kong X J, Wu Y, Long L S, et al. J. Am. Chem. Soc., 2009, 131(20):6918-6919 doi: 10.1021/ja901214d
Kong X J, Long L S, Zheng Z, et al. Acc. Chem. Res., 2009, 43(2):201-209
Zheng Y Z, Evangelisti M, Tuna F, et al. J. Am. Chem. Soc., 2012, 134(2):1057-1065 doi: 10.1021/ja208367k
Peng J B, Zhang Q C, Kong X J, et al. J. Am. Chem. Soc., 2012, 134(7):3314-3317 doi: 10.1021/ja209752z
Zheng Y Z, Evangelisti M, Winpenny R E P. Chem. Sci., 2011, 2(1):99-102 doi: 10.1039/C0SC00371A
Abtab S M T, Majee M C, Maity M, et al. Inorg. Chem., 2014, 53(3):1295-1306 doi: 10.1021/ic401484d
Ungur L, Thewissen M, Costes J P, et al. Inorg. Chem., 2013, 52(11):6328-6337 doi: 10.1021/ic302568x
Li J, Wei R M, Pu T C, et al. Inorg. Chem. Front., 2017, 4(1):114-122 doi: 10.1039/C6QI00407E
Liu J L, Wu J Y, Huang G Z, et al. Sci. Rep., 2015, 5:16621 doi: 10.1038/srep16621
Mondal K C, Sundt A, Lan Y, et al. Angew. Chem., Int. Ed., 2012, 51(30):7550-7554 doi: 10.1002/anie.201201478
Ferguson A, Lawrence J, Parkin A, et al. Dalton Trans., 2008(45):6409-6414 doi: 10.1039/b807447j
Ako A M, Mereacre V, Clérac R, et al. Inorg. Chem., 2009, 48(14):6713-6723 doi: 10.1021/ic900645d
Peng Y, Tian C B, Lan Y H, et al. Eur. J. Inorg Chem., 2013, 2013(32):5534-5540 doi: 10.1002/ejic.v2013.32
Sheldrick G M. SHELXS-97, Program for the Solution of Crystal Sturcture, University of Göttingen, Germany, 1997.
Sheldrick G M. SHELXL-97, Program for the Refinement of Crystal Sturcture, University of Göttingen, Germany, 1997.
任艳秋, 韩伟, 程美令, 等.无机化学学报, 2014, 30(11):2635-2644 http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=wjhxxb201411026REN Yan-Qiu, HAN Wei, CHENG Mei-Ling, et al. Chinese J. Inorg. Chem., 2014, 30(11):2635-2644 http://www.wanfangdata.com.cn/details/detail.do?_type=perio&id=wjhxxb201411026

Figure 1 Molecular structure of 1
Hydrogen atoms, free Cl- and solvent molecules are omitted for clarity

Scheme 1 Different coordination modes of L and HL
a, b and c: μ3-η1: η2: η3; d: μ3-η1: η1: η3
Table 1. Crystal data and structure refinement for 1
| Empirical formula | C42H110Cl4Co8Dy2N8O34 |
| Formula weight | 2 201.55 |
| Crystal size/mm | 0.15×0.14×0.12 |
| Temperature/K | 296(2) |
| Crystal system | Triclinic |
| space group | P1 |
| a/nm | 1.160 4(8) |
| b/nm | 1.322 7(9) |
| c/nm | 1.465 3(9) |
| α/(°) | 72.181(7) |
| β/(°) | 67.638(7) |
| γ/(°) | 65.939(7) |
| V/nm3 | 1.869(2) |
| Z | 1 |
| Dc/(g·cm-3) | 1.956 |
| μ(Mo Kα)/mm-1 | 3.928 |
| F(000) | 1 098 |
| θ range/(°) | 2.491~25.997 |
| Reflection collected | 28 782 |
| Independent reflection (Rint) | 7 318 (0.051 1) |
| Data, restrain, parameter | 7 318, 38, 442 |
| Limiting indices (h, k, l) | -14~14, -16~16, -18~18 |
| R1, wR2 [I > 2σ(I)] | 0.056 1, 0.153 1 |
| R1, wR2 (all data) | 0.077 7, 0.199 6 |
| S on F2 | 1.147 |
| Largest diff. peak and hole/(e·nm-3) | 3 700 and -2 392 |
 下载: 导出CSV
下载: 导出CSV
Table 2. Selected bond lengths (nm) and bond angles (°) of 1
| Dy(1)-O(9) | 0.229 7(5) | Co(3)-O(4) | 0.188 0(6) | Co(2)-O(10) | 0.194 1(6) |
| Dy(1)-O(12) | 0.237 8(6) | Co(3)-N(2) | 0.192 8(9) | Co(2)-N(3) | 0.196 3(8) |
| Dy(1)-O(7) | 0.240 3(8) | Co(1)-O(9) | 0.205 8(6) | Co(4)-O(2) | 0.206 0(6) |
| Dy(1)-O(4) | 0.240 4(6) | Co(1)-O(8) | 0.208 8(7) | Co(4)-Cl(1) | 0.236 9(4) |
| O(9)-Dy(1)-O(12) | 125.6(2) | O(9)-Co(1)-O(8) | 94.9(3) | N(2)-Co(3)-N(1) | 100.0(4) |
| O(9)-Dy(1)-O(7) | 76.4(2) | O(12)-Co(2)-O(11) | 177.3(3) | O(2)-Co(4)-O(6) | 150.0(3) |
| O(12)-Dy(1)-O(7) | 119.5(3) | O(4)-Co(3)-O(2) | 175.5(3) | ||
| O(7)-Dy(1)-O(4) | 72.6(2) | O(4)-Co(3)-N(2) | 86.6(3) |
下载: 导出CSV
Table 3. Hydrogen bond parameters of 1
| D-H…A | d(D-H)/nm | d(H…A)/nm | d(D…A)/nm | ∠DHA/(°) |
| O(1W)-H(1WA)…O(11)#1 | 0.096 | 0.209 | 0.270 5(11) | 120.3 |
| O(16)-H(16C)…O(1W)#2 | 0.096 | 0.192 | 0.272 8(17) | 140.2 |
| O(15)-H(15C)…N(3)#3 | 0.096 | 0.222 | 0.295 0(13) | 132.4 |
| C(7)-H(7B)…O(1W)#4 | 0.097 | 0.261 | 0.351 1(19) | 154.7 |
| C(21)-H(21A)…Cl(2)#2 | 0.096 | 0.269 | 0.360(2) | 157.3 |
| C(4)-H(4A)…Cl(1) | 0.097 | 0.292 | 0.359 6(12) | 127.3 |
| C(6)-H(6A)…O(1) | 0.097 | 0.26 | 0.319 3(15) | 119.8 |
| C(16)-H(16B)…O(4) | 0.097 | 0.253 | 0.313 8(12) | 120.5 |
| C(14)-H(14A)…Cl(2)#1 | 0.097 | 0.267 | 0.361 9(12) | 166.8 |
| C(9)-H(9A)…O(7) | 0.097 | 0.249 | 0.307 1(13) | 118.2 |
| C(3)-H(3B)…Cl(2)#4 | 0.097 | 0.291 | 0.381 7(14) | 156.8 |
| C(12)-H(12A)…O(14)#1 | 0.097 | 0.265 | 0.330 7(12) | 125.4 |
| O(1)_H(1C)…O(1W) | 0.085 | 0.196 | 0.262 1(11) | 133.4 |
| O(9)-H(9C)…O(11)#1 | 0.098 | 0.175 | 0.262 5(8) | 146.6 |
| Symmetry transformations used to generate equivalent atoms: #1: -x+1, -y+1, -z+1; #2: x+1, y, z; #3: x, y, z+1; #4: -x+1, -y, -z+1. | ||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们







 下载:
下载: