
图 1
不同比例的镁掺杂羟基磷灰石的TEM图
Figure 1.
TEM images of Mg-HA particles synthesized at different feeding molar ratios

羟基磷灰石(Ca10(PO4)6(OH)2,Hydroxyapatite, HA), 与人体内骨组织和牙齿等矿化组织中的无机质在化学成分和结构上相似[1-4],具有良好的生物活性和生物相容性[5-7],可作为药物载体用于药物的负载和释放[8-10],在释放药物后可被人体降解吸收或全部随粪便排出,被认为是具有发展前景的生物活性材料之一。羟基磷灰石可被其他阴阳离子替换,通过掺杂不同的金属元素(如Mg2+、Cu2+、Sr2+、Zn2+等)或阴离子(CO32-、F-等)可使羟基磷灰石具有不同的形貌[2, 11-12],进而影响其稳定性、比表面积和对药物的负载能力等[13-14]。
HA具有很好的生物相容性和生物安全性。同时,它的化学稳定性很好,对许多药物都不起化学反应,可以作为多种药物的缓释载体使用。HA一般为纳米实心棒状结构,不像碳纳米管具有空腔结构,载药量不够高(低于10%),因此,人们采用与其它材料复合如壳聚糖或制备成微球结构来解决上述问题[15-16]。但对直接从合成途径设计对HA纳米棒结构进一步改性,实现比表面面积的有效增加目前报道的还较少。镁(Mg)与钙(Ca)元素同属碱土金属即元素周期表中ⅡA族元素,化学性质非常相近。Mg是人体必须的微量元素之一,对人体起着十分重要的作用,可参与催化人体内200多个酶生物反应过程,并且Mg在维持骨骼细胞结构结合骨骼韧性方面起了重要作用[17-18]。Mg取代羟基磷灰石中Ca的位置后,会使羟基磷灰石的晶体结构发生变化,从而使羟基磷灰石的形貌和理化性质发生改变[19]。近年来,已有文献报道多种Mg掺杂的羟基磷灰石的合成方法,这些研究表明,Mg的掺杂对于羟基磷灰石的生物活性有明显的提高。例如,Kalita[20]将氧化镁和氧化锌作为掺杂剂加入到羟基磷灰石前驱体粉末中,通过高温固相合成了Mg/Zn-HA,其力学性能与生物活性均优于单纯的HA;Kim[21]利用化学共沉淀法合成了Si/Mg-HA,具有良好的生物活性,可用于骨填充材料。但是由于Mg与Ca的离子半径与亲水性都相似,会引起离子竞争效应,降低产物的结晶度[22-23]。
本文拟利用与钙元素化学和生物学性能相近的镁元素掺杂HA, Ca10(PO4)6(OH)2分子结构中Ca部分被Mg取代形成MgxCa10-x(PO4)6(OH)2复合物,能在HA纳米棒状结构上形成缺陷,从而增大HA比表面积和提高生物化学活性,对于载药能力的有效提高和药物的缓释能起重要作用。研究通过逐步化学沉淀反应和Mg2+、Ca2+离子置换反应一锅法制备了一系列不同含量镁掺杂的纳米羟基磷灰石(Mg-HA)。将以Mg(NO3)2·6H2O和(NH4)2HPO4发生反应形成纳米羟基磷酸镁为前驱体,进一步滴加Ca(NO3)2·4H2O和(NH4)2HPO4溶液最终形成Mg-HA。对其纳米粉体的理化特性、生物学性能进行表征,选择化疗药物顺铂(Cisplatin,cDDP)作为模型药物,评价新型Mg-HA纳米材料载药特性及其在药物载体方面的应用前景。
四水合硝酸钙(Ca(NO3)2·4H2O,AR),六水合硝酸镁(Mg(NO3)2·6H2O,AR),磷酸氢二铵((NH4)2HPO4,AR),二甲亚砜(C2H6SO,dimethyl sulfoxide,DMSO)均购于国药集团化学试剂有限公司;浓氨水(含量(以NH3计)25%~28%,AR)购于上海联试有限公司;1, 1, 1-三羟甲基乙烷(1, 1, 1-tris(hydroxymethyl)ethane,TME,97%),顺铂(cis-diammineplatinum(Ⅱ) dichloride, cDDP,Pt 65%)均购于Alfa Aesar;胎牛血清(FBS)、DMEM干粉培养基(高糖)、青-链霉素、0.25%胰蛋白酶-0.02%EDTA消化液、非必需氨基酸以及磷酸盐缓冲液(PBS)、3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT试剂)均购于Sigma-Aldrich。
通过逐步化学沉淀反应一锅法制备镁掺杂的纳米羟基磷灰石。250 mL烧瓶中加入10 mL浓度为33.4 mmol· L-1的Mg(NO3)2·6H2O溶液,加入1%(w/w)TME,磁力搅拌条件下加热至85 ℃,缓慢滴加10 mL浓度为20 mmol· L-1的(NH4)2HPO4溶液。用浓氨水调节反应溶液的pH值为8.5左右。保持反应3 h后,向体系中滴加物质的量浓度为33.4 mmol· L-1的Ca(NO3)2·4H2O溶液,其中Ca与Mg的物质的量比控制为1:1、2.5:1、5:1和7.5:1,反应3 h后再向体系中滴加浓度为20 mmol· L-1的(NH4)2HPO4溶液, 保证(nCa+nMg)/nP=1.67。反应保持24 h,陈化12 h以上,用超纯水去除多余的TME,冷冻干燥后得到Mg-HA。
利用透射电镜(TEM,Tecnai G2 Spirit型,美国FEI公司)观测样品的形态和大小,工作电压为120 kV。利用傅里叶变换红外光谱仪(FTIR,Nicolet 6700型,美国Nicolet公司)表征其化学结构;样品用溴化钾(KBr)压片法制样,透射模式检测,检测范围为4 000~400 cm-1。利用X射线衍射仪(XRD,D/max-2200/PC型,日本Rigaku公司)表征物相结构,检测条件为铜靶(λ=0.154 05 nm),电流20 mA,电压40 kV,扫描速率0.02°·s-1,扫描范围:2θ=5°~80°。样品的比表面积和孔隙度通过比表面积孔隙度及化学吸附分析仪(ASAP 2010 M+C型,美国Micromeritics公司)检测;样品经过100 ℃除湿处理后,以Brunauer-Emmett-Teller (BET)模型分析计算样品的比表面积数据,并通过脱附等温线计算得到孔结构的数据。利用电感耦合等离子光谱发射光谱仪(ICP,iCAP6300型,美国Thermo Fisher Scientific公司)定量分析样品中Ca、Mg和P元素的含量;样品的热稳定性通过热重分析仪(TGA,Pyris1型,美国PerkinElmer仪器公司)检测;称取一定量的样品,置于氧化铝坩埚中,N2气氛,检测温度范围为40~900 ℃,升温速率为20 ℃·min-1。
称取10 mg Mg-HA(nCa:nMg=1:1、2.5:1、5:1和7.5:1)加入到5 mL PBS(pH=7.4)中超声均匀分散。称取15 mg顺铂(cDDP)溶于DMSO与PBS混合溶液中(VPBS:VDMSO=4),逐滴滴入Mg-HA分散液中,在37 ℃条件下保持24 h使顺铂充分吸附在Mg-HA上;然后,离心分离、冷冻干燥后得到装载顺铂的Mg-HA。用ICP测定铂元素含量计算出上清液中未被吸附顺铂的浓度C1,已知初始的顺铂浓度为C0,溶液体积为V,载药样品总质量为m,装载药物质量为m0,则样品的载药量通过以下公式计算得出:
|
$ {\rm{DLC}} = ({m_0}/m)\times 100\% = [({C_0} - {C_1})V/m]\times 100\% $ |
药物缓释效果测定:称取4 mg Mg-HA/cDDP样分散在2 mL PBS (pH=5.5或pH=7.4)中,并置于透析袋中(截留分子量为1 kDa)。将透析袋置于30 mL pH=5.5或7.4的PBS(含2 mL DMSO助溶)中(37 ℃,12 000 r·min-1),分别在0.25、0.5、1、2、4、8、12、24、48和72 h时取样ICP检测缓释药物,将透析袋外液体全部移除,重新加入30 mL PBS(含2 mL DMSO助溶),(pH=5.5或pH=7.4)。
载药量与释药量测定数据是3组平行实验结果的平均值。
实验选用的L929 (小鼠成纤维细胞)细胞系与HeLa(人宫颈癌细胞)细胞系购于中科院上海细胞库。其培养方法如下:用含有10%(V/V)胎牛血清(FBS)、100 units·m L-1链霉素、100 units·m L-1青霉素的DMEM培养基进行细胞培养,置于75 cm2的培养瓶中贴壁生长。培养瓶置于温度为37 ℃,含有5% CO2(V/V)的培养箱中培养,每隔2~3 d细胞进行传代或实验操作。
取对数生长期的L929细胞胰酶消化制成细胞悬液,使用DMEM培养基对细胞进行稀释,细胞密度约为每毫升5×104个,小心接种于96孔板,每孔中加入200 μL细胞悬液,使每孔中含有约1×104个细胞。培养24 h,弃培养液,每孔加入50 μL一系列不同浓度的Mg-HA(PBS配制), 浓度依次为0.3、0.6、1.25、2.5、5、10、20、40和80 μg·m L-1,每个浓度设置5个复孔,对照组加入等体积PBS,置于37 ℃、含有5%(V/V) CO2的培养箱中培养24、48和72 h后,吸出培养液,再加入PBS小心清洗96孔板2次后,每孔加入200 μL新鲜的DMEM培养基,然后加入用PBS配制的浓度为5 mg·m L-1的MTT溶液20 μL,置于37 ℃培养箱中继续孵育4 h,然后弃去培养液,每孔加入150 μL DMSO,振板10 min,通过酶标仪测490 nm波长的吸光度值。每组有6个平行孔(n=6),实验共重复3次。
按1.6所述方法于96孔板中接种细胞培养24 h,分别加入50 μL Mg-HA/cDDP溶液,梯度浓度以顺铂浓度计算,设为0.3、0.6、1.25、2.5、5、10、20、40和80 μg·m L-1,每个浓度设置5个复孔,对照组加入等体积PBS,置于37 ℃、含有5%(V/V) CO2的培养箱中培养24、48和72 h后,弃培养液,再用PBS小心清洗96孔板2次后,每孔加入200 μL新鲜的DMEM培养基,再加入用PBS配制的浓度为5 mg·m L-1的MTT溶液20 μL,置于培养箱中继续孵育4 h,然后弃去培养液,每孔加入150 μL DMSO,振板10 min,通过酶标仪测每孔在490 nm波长下的光密度值(Optical density,OD),OD实表示实验组OD平均值,OD空表示空白组OD平均值,根据公式计算细胞生长抑制率(η)。
|
$ \eta = (1 - {\rm{O}}{{\rm{D}}_{实}}/{\rm{O}}{{\rm{D}}_{空}})\times 100\% $ |
对照组为单独作用顺铂对HeLa细胞的生长抑制率。每组有6个平行孔(n=6),实验共重复3次。
通过TEM能清楚观察到不同投料物质的量比制得的镁掺杂羟基磷灰石(nCa:nMg=1:1、2.5:1、5:1和7.5:1)的形貌和尺寸大小,以及镁掺杂量对纳米颗粒形态和尺寸的影响(图 1)。如图 1a所示,羟基磷灰石的形貌为长500~600 nm,直径为50~100 nm的棒状结构。掺杂镁后的羟基磷灰石在形貌上与羟基磷灰石相比发生了较大变化(如图 1(b~e)),产物的尺寸为300~400 nm,直径约为100~200 nm,外观上有束状纳米纤维,呈稻草状结构,在更高的放大倍数下(图 1d′),可以观察到其束状形态是由许多直径10 nm左右的定向排列纳米纤维组成;电子衍射图谱中的衍射环清楚地表明其为各向异性的多晶结构。当投料比nCa:nMg=1:1 (图 1b),2.5:1(图 1c)时,其纳米纤维并不明显,随着Mg比例的减小,当投料比nCa:nMg=5:1(图 1d),7.5:1(图 1e)时产物外观上的纤维状结构趋于明显,并且产物形貌的均一性较好。
根据不同投料物质的量比所制得的Mg-HA的XRD衍射峰(图 2)与纯羟基磷灰石的衍射峰相符合,并且随着Mg在产物中的比例增加,产物的晶型的混乱度也有所增加,与文献报道的规律相符合[25]。在投料物质的量比为2.5:1时,可以明显看到有杂质峰的存在,为Mg3(PO3)2·5H2O前体导致[26],说明在投料比为2.5:1时,有部分Mg离子在被Ca离子替换出来后,未能继续参与反应。其他不同投料比的条件下,由于杂质含量较低,并不能明显观测到杂质峰。

图 3是不同比例的Mg-HA的红外吸收谱图。谱图中2 800~3 600 cm-1的宽峰以及1 635 cm-1左右的吸收峰是产物中的羟基或吸收空气中的水的羟基O-H键振动吸收峰。1 430 cm-1处的峰是羰基C=O的吸收峰,表明了样品在制备过程中,空气中的CO2可能参与了反应,形成了碳酸根离子(CO32-)。927~1 160 cm-1处的单峰是由磷酸根离子(PO43-)中P-O的ν3伸缩振动造成的。497~608 cm-1处的双峰则是由于磷酸(PO43-)的ν4弯曲振动造成的[27]。

表 1是不同比例的镁掺杂羟基磷灰石的比表面积数据,其中羟基磷灰石的比表面积为43 m2· g-1,而不同投料物质的量比的镁掺杂羟基磷灰石的比表面积均大于羟基磷灰石,约为2~3倍,这可能是受到其表面的大量纳米纤维结构的影响,极大的增大了镁掺杂羟基磷灰石的比表面积。
 下载:
导出CSV
下载:
导出CSV
| nCa:nMg | BET surface area / (m2·g-1) | Average pore diameter / nm | Pore volume / (cm3·g-1) |
| 1:1 | 99 | 10.1 | 0.26 |
| 2.5:1 | 116 | 10.5 | 0.31 |
| 5:1 | 103 | 9.8 | 0.35 |
| 7.5:1 | 100 | 11.2 | 0.29 |
| HA | 43 | 22.3 | 0.24 |
为了验证最终产物中的Ca与Mg的含量是否与实际投料比相一致,用ICP来定量检测样品中Ca、Mg和P。结果如表 2所示。所有产物中nCa:nMg的实际比值均大于投料比,分别为6.94,3.64,7.90,9.56(其对应的nCa:nMg=1:1、2.5:1、5:1和7.5:1)。说明在反应过程中,被钙离子置换出来的镁离子有部分未能重新参与反应。当nCa:nMg=5:1,7.5:1时Ca:Mg的实际比值较为接近。羟基磷灰石的nCa/nP的理论值为1.67,当nCa:nMg=5:1,7.5:1时,检测出的(nCa+nMg)/nP的数值均接近于理论值,分别为1.64,1.67,此时产物中的实际镁含量分别为2.26%和2.17%(w/w)。
下载:
导出CSV
| nCa:nMg | Measured mass fraction / % | Measured molar ratios | Chemical formula (regardless of other impurities) |
||||
| Ca | Mg | P | nCa /nMg | (nCa+nMg)/nP | |||
| 1:1 | 28.58 | 2.47 | 15.84 | 6.94 | 1.6 | Ca8.4Mg1.2(PO4)6(OH)2 | |
| 2.5:1 | 25.12 | 4.14 | 16.11 | 3.64 | 1.54 | Ca7.2Mg2(PO4)6(OH)2 | |
| 5:1 | 29.86 | 2.26 | 15.89 | 7.9 | 1.64 | Ca8.7Mg1.1(PO4)6(OH)2 | |
| 7.5:1 | 33.96 | 2.17 | 17.27 | 9.56 | 1.67 | Ca9.1Mg0.9(PO4)6(OH)2 | |
| HA | 38.1 | — | 17.65 | — | 1.67 | Ca10(PO4)6(OH)2 | |
通过不同投料比制得的Mg-HA(nCa:nMg=1:1、2.5 :1、5:1和7.5:1)的热稳定性热失重曲线如图 4所示,随着Mg在产物中的比例增加,样品的热稳定性也逐渐增加。在30~800 ℃的升温范围内,Mg-HA和HA的失重主要为表面吸附的水失重(0~200 ℃,体中的晶格水失重(200~400 ℃)和羟基磷灰石中羟基的失重(600~800 ℃)[28]。所制得的Mg-HA的热稳定性均较好,在30~800 ℃的升温范围内,失重均小于10%。

上述实验结果表明,通过逐步反应一锅法可以获得这种形貌尺寸可控的镁掺杂的纳米羟基磷灰石。其离子置换的过程可能为:
|
$ \begin{array}{l} {\rm{M}}{{\rm{g}}_3}{({\rm{P}}{{\rm{O}}_4})_2}\cdot5{{\rm{H}}_2}{\rm{O}}\left( {\rm{s}}\right) + 3{\rm{C}}{{\rm{a}}^{2 + }}\to\\ \quad\quad\quad\quad 3{\rm{M}}{{\rm{g}}^{2 + }} + {\rm{C}}{{\rm{a}}_3}{({\rm{P}}{{\rm{O}}_4})_2}\left( {\rm{s}}\right) + 5{{\rm{H}}_2}{\rm{O}} \end{array} $ |
|
$ \begin{array}{l} x{\rm{C}}{{\rm{a}}_3}{({\rm{P}}{{\rm{O}}_4})_2}\left( {\rm{s}}\right) +\left( {30 - 3x}\right){\rm{M}}{{\rm{g}}^{2 + }} +\left( {18 - 2x}\right){\rm{HP}}{{\rm{O}}_4}^{2 - } + 6{{\rm{H}}_2}{\rm{O}}\to\\ \quad\quad\quad\quad 3{\rm{C}}{{\rm{a}}_x}{\rm{M}}{{\rm{g}}_{10 - x}}({\rm{P}}{{\rm{O}}_4})6{\left( {{\rm{OH}}}\right)_2}\left( {\rm{s}}\right) +\left( {24 - 2x}\right){{\rm{H}}^ + }\\ \\ \end{array} $ |
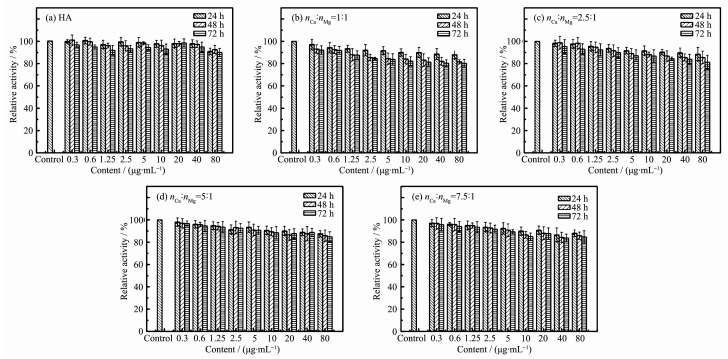
通过MTT法测定不同Ca和Mg的投料比制得的Mg-HA对L929细胞系的体外细胞毒性。结果如图 5所示,随着Mg-HA浓度的增加和孵育时间的延长,材料对L929细胞系的毒性略有增加,但最终细胞存活率都保持在80%以上,结果表明所制得的镁掺杂的纳米羟基磷灰石对于L929细胞系的体外细胞毒性较低,具有较好的生物相容性。
不同投料比制得的Mg-HA(nCa:nMg=1:1、2.5:1、5:1和7.5:1)的载药量随着镁的比例的增加,载药量有升高的趋势,这与材料上的纤维状的结构及其比表面积有关(图 6)。其中(nCa:nMg=5:1的Mg-HA载药量最高,为54.84%,所合成的Mg-HA的载药量均大于羟基磷灰石HA的载药量。

羟基磷灰石对于顺铂药物的负载主要为吸附原理,Kawasaki[29]认为羟基磷灰石表面有2种吸附位置:① Ca位置:当羟基磷灰石表面为OH-时,该位置连着2个Ca2+,当在水溶液中,OH-至少在一瞬间空缺,该位置便形成了1个可吸附阴离子如PO43-、羧基等的吸附位置;② P位置:当羟基磷灰石表面为Ca原子时,该位置周围连着6个O原子,在水溶液中,表面的Ca位置至少在一瞬间空缺,该位置便形成了1个可吸附阳离子如Sr2+、K+等的吸附位置。因此,在羟基磷灰石负载顺铂时,主要原理为顺铂中心的二价铂通过该吸附原理吸附在羟基磷灰石的P位置。掺杂镁元素后,会导致羟基磷灰石的结构缺陷,更有利于顺铂的吸附负载。
综合上述物理化学性能表征的结果,以及载药量的测试结果,我们选定投料比nCa:nMg=5:1制备的镁掺杂羟基磷灰石(实际镁含量为2.26%(w/w))作为研究对象进行后续的实验研究。
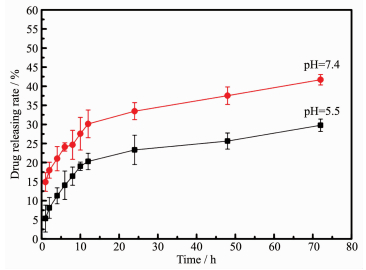
图 7为在pH=5.5和pH=7.4条件下测定Mg-HA/cDDP的药物缓释曲线。72 h后材料在pH=7.4和5.5时顺铂药物累积释放量分别达到29.77%和41.72%(w/w)。表明镁掺杂羟基磷灰石对顺铂有一定的缓释效果,在pH=5.5条件下药物累积释放量高于pH=7.4时,表明该负载药物后的材料在肿瘤部位的微酸环境[30-32]中能有较好的药物缓释结果。
如图 8所示,Mg-HA (nCa:nMg=5:1,实际镁含量为2.26%)负载顺铂药物后对HeLa细胞有明显的生长抑制作用,随着作用时间的增长,顺铂浓度的增加,对HeLa细胞增殖抑制作用也在增加。80 μg·m L-1载cDDP药物的Mg-HA (以cDDP计)与细胞共孵育24和48 h后,细胞生长的抑制率分别为80.46%,87.79%。证明Mg-HA负载顺铂后能具有很好抑制癌细胞生长的效果。

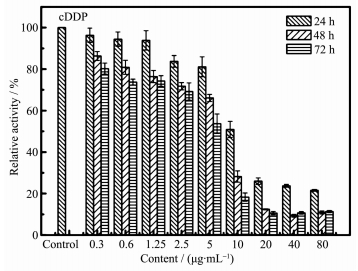
如图 9所示,单独作用顺铂对HeLa细胞有生长抑制作用。随着作用时间的增长以及顺铂浓度的增加,对HeLa细胞的增殖抑制作用也变得显著。80 μg·m L-1 cDDP药物与细胞共孵育24、48和72 h后,细胞生长的抑制率分别为78.43%、89.11%和88.65%。实验结果与利用Mg-HA/cDDP作用于HeLa细胞对其的生长抑制率相一致,证明所制得的Mg-HA作为药物载体不会影响抗癌药物的药效。
本研究使用了逐步反应步骤一锅法合成了镁掺杂的纳米羟基磷灰石,通过调节Ca和Mg的投料比,能有效地调控Mg-HA的形貌和尺寸,其中当nCa:nMg=5:1时,产物的形貌均一性较好,比表面积较大,有利于药物的负载,此时所制得的Mg-HA中Mg的实际含量为2.26%。体外细胞毒性实验表明所制备材料的体外细胞毒性较低,具有良好的生物相容性。选择nCa:nMg=5:1的Mg-HA作为药物载体负载抗癌药物顺铂,载药率为54.84%,载药性能比HA有了较大提升,且在pH=5.5时,能有较好的药物缓释效果。材料负载药物后仍能对HeLa细胞有较好的生长抑制效果,80 μg·m L-1药物与细胞共孵育24和48 h后,细胞生长的抑制率分别为80.46%和87.79%,其结果与相同浓度与孵育时间条件下,单独顺铂药物与HeLa细胞作用的结果相一致。本文制备的镁掺杂的纳米羟基磷灰石能够作为一种安全的药物载体在生物医药领域等具有较好的应用前景。
Iijima M, Moriwaki Y, Yamaguchi R, et al. Connect. Tissue Res., 1997, 36(2):73-83 doi: 10.3109/03008209709160215
Vallet-Regí M, González-Calbet J M. Prog. Solid State Chem., 2004, 32(1):1-31 doi: 10.1016/j.progsolidstchem.2004.07.001
Wang L, Nancollas G H. Chem. Rev., 2008, 108(11):4628-4669 doi: 10.1021/cr0782574
Dorozhkin S V. Biomaterials, 2010, 31(7):1465-1485 doi: 10.1016/j.biomaterials.2009.11.050
Zhao J, Duan K, Zhang J W, et al. Mater. Sci. Eng., C, 2011, 31(3):697-701 doi: 10.1016/j.msec.2010.12.011
Fathi M H, Hanifi A, Mortazavi V. J. Mater. Process. Technol., 2008, 202(1/2/3):536-542 doi: 10.1016/j.jmatprotec.2007.10.004
Abdulkareem E H, Memarzadeh K, Allaker R P, et al. J. Dent., 2015, 43(12):1462-1469 doi: 10.1016/j.jdent.2015.10.010
Ye F, Guo H, Zhang H, et al. Acta Biomater., 2010, 6(6):2212-2218 doi: 10.1016/j.actbio.2009.12.014
Sun R, Chen K, Lu Y. Mater. Res. Bull., 2009, 44(10):1939-1942 doi: 10.1016/j.materresbull.2009.06.015
Zhao Q F, Wang T Y, Wang J, et al. Appl. Surf. Sci., 2011, 257(23):10126-10133 doi: 10.1016/j.apsusc.2011.06.161
Gomes S, Nedelec J M, Jallot E, et al. Chem. Mater., 2011, 23(12):3072-3085 doi: 10.1021/cm200537v
Murata H, Shitara K, Tanaka I, et al. J. Phys.:Condens. Matter, 2010, 22(38):384213-384218 doi: 10.1088/0953-8984/22/38/384213
Evis Z, Webster T J. Adv. Appl. Ceram., 2011, 110(5):311-320 doi: 10.1179/1743676110Y.0000000005
Webster T J, Massa-Schlueter E A, Smith J L, et al. Biomaterials, 2004, 25(11):2111-2121 doi: 10.1016/j.biomaterials.2003.09.001
赵宏霞, 金花, 蔡继业.无机化学学报, 2010, 26(1):106-111 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20100120&journal_id=wjhxxbcnZHAO Hong-Xia, JIN Hua, CAI Ji-Ye. Chinese J. Inorg. Chem., 2010, 26(1):106-111 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20100120&journal_id=wjhxxbcn
姚爱华, 徐为, 艾凡荣, 等.无机材料学报, 2011, 26(9):974-978 http://www.jim.org.cn/CN/abstract/abstract12240.shtmlYAO Ai-Hua, XU Wei, AI Fan Rong, et al. J. Inorg. Mater., 2011, 26(9):974-978 http://www.jim.org.cn/CN/abstract/abstract12240.shtml
Niki Y, Matsumoto H, Suda Y, et al. Biomaterials, 2003, 24(8):1447-1457 doi: 10.1016/S0142-9612(02)00531-8
Kalita S J, Bhatt H A. Mater. Sci. Eng., C, 2007, 27(4):837-848 doi: 10.1016/j.msec.2006.09.036
Suchanek W L, Byrappa K, Shuk P, et al. Biomaterials, 2004, 25(19):4647-4657 doi: 10.1016/j.biomaterials.2003.12.008
Kim S R, Lee J H, Kim Y T, et al. Biomaterials, 2003, 24(8):1389-1398 doi: 10.1016/S0142-9612(02)00523-9
Landi E, Logroscino G, Proietti L, et al. J. Mater. Sci.-Mater. Med., 2008, 19(1):239-247 doi: 10.1007/s10856-006-0032-y
Bigi A, Falini G, Foresti E, et al. J. Inorg. Biochem., 1993, 49(1):69-78 doi: 10.1016/0162-0134(93)80049-F
Wan Y Z, Huang Y, He F, et al. Surf. Coat. Technol., 2006, 201(6):2904-2909 doi: 10.1016/j.surfcoat.2006.06.004
Zhang X J, Lin D Y, Yan X H, et al. J. Cryst. Growth, 2011, 336(1):60-66 doi: 10.1016/j.jcrysgro.2011.09.039
Ren F, Leng Y, Xin R, et al. Acta Biomater., 2010, 6(7):2787-2796 doi: 10.1016/j.actbio.2009.12.044
Yuan X Y, Zhu B S, Tong G S, et al. J. Mater. Chem. B, 2013, 1(47):6551-6559 doi: 10.1039/c3tb21315c
Eslami H, Solati-Hashjin M, Tahriri M. Journal of Ceramic Processing Research, 2008, 9(3):224-229
Ren F, Leng Y R, Ge X. Acta Biomater., 2010, 6(7):2787-2796 doi: 10.1016/j.actbio.2009.12.044
Kawasaki T, Niikura M, Kobayashi Y. J. Chromatogr. A, 1990, 515:125-148 doi: 10.1016/S0021-9673(01)89307-9
Ulbrich K, Etrych T, Chytil P, et al. Adv. Drug Delivery Rev., 2004, 277(1/2):63-72
Kratz F, Beyer U, Schütte M T. Crit. Rev. Ther. Drug Carrier Syst., 1999, 16(3):245-248 doi: 10.1615/CritRevTherDrugCarrierSyst.v16.i3
Rodrigues P C, Roth T, Fiebig H, et al. Bioorg. Med. Chem., 2006, 14(12):4110-4117 doi: 10.1016/j.bmc.2006.02.007

图 1 不同比例的镁掺杂羟基磷灰石的TEM图
Figure 1 TEM images of Mg-HA particles synthesized at different feeding molar ratios
Inset: corresponding electron diffraction pattern

图 2 不同比例的镁掺杂羟基磷灰石的XRD图
Figure 2 XRD patterns of Mg-HA materials synthesized at different feeding molar ratios

图 3 不同比例的镁掺杂羟基磷灰石的FTIR图谱
Figure 3 FTIR spectra of the Mg-HA materials synthesized at different feeding molar ratios

图 4 TGA分析不同比例的镁掺杂羟基磷灰石的热稳定性
Figure 4 Thermal stability of the Mg-HA materials synthesized at different feeding molar ratios

图 5 不同投料比制得的镁掺杂羟基磷灰石以不同浓度梯度与L929细胞共同培养不同时间后相对细胞活性的数据图
Figure 5 Relative cell activity of L929 cells against Mg-HAs synthesized at various feeding molar ratios compared with HA at different concentrations with different times
(a) HA; (b) 1:1; (c) 2.5:1; (d) 5:1; (e) 7.5:1; Data were collected after 24, 48 and 72 h (n=6)

图 6 不同比例的镁掺杂羟基磷灰石对顺铂药物的载药量
Figure 6 Drug loading rate of Mg-HA materials synthesized at different feeding molar ratios (nCa:nMg) with cDDP in comparison with HA

图 7 镁掺杂羟基磷灰石(含2.26%镁元素)在pH=5.5和7.4时的药物缓释曲线
Figure 7 Drug releasing rate of Mg-HA (with 2.26% Mg) materials with cDDP at pH=5.5 and 7.4

图 8 镁掺杂羟基磷灰石(含2.26%镁元素)载顺铂后以不同浓度梯度(以顺铂计)与HeLa细胞共同培养后相对细胞活性的数据图
Figure 8 Relative cell activity of HeLa cells against Mg-HA/ cDDP (with 2.26% Mg) at different concentrations for cDDP
Data were collected after 24 h, 48 h (n=6)

图 9 顺铂(cDDP)以不同浓度梯度与HeLa细胞共同培养后相对细胞活性的数据图
Figure 9 Relative cell activity of HeLa cells against cDDP at different concentrations
Data were collected after 24, 48 and 72 h (n=6)
表 1 不同比例的镁掺杂羟基磷灰石的比表面积、孔径、孔容数据
Table 1. Specific surface area, pore volume, and average pore diameter of Mg-HA materials synthesized at different feeding molar ratios
| nCa:nMg | BET surface area / (m2·g-1) | Average pore diameter / nm | Pore volume / (cm3·g-1) |
| 1:1 | 99 | 10.1 | 0.26 |
| 2.5:1 | 116 | 10.5 | 0.31 |
| 5:1 | 103 | 9.8 | 0.35 |
| 7.5:1 | 100 | 11.2 | 0.29 |
| HA | 43 | 22.3 | 0.24 |
 下载: 导出CSV
下载: 导出CSV
表 2 不同比例的镁掺杂羟基磷灰石的元素Ca、Mg、P组成分析
Table 2. Chemical analysis of Ca, Mg and P in Mg-HA materials synthesized at different feeding molar ratios
| nCa:nMg | Measured mass fraction / % | Measured molar ratios | Chemical formula (regardless of other impurities) |
||||
| Ca | Mg | P | nCa /nMg | (nCa+nMg)/nP | |||
| 1:1 | 28.58 | 2.47 | 15.84 | 6.94 | 1.6 | Ca8.4Mg1.2(PO4)6(OH)2 | |
| 2.5:1 | 25.12 | 4.14 | 16.11 | 3.64 | 1.54 | Ca7.2Mg2(PO4)6(OH)2 | |
| 5:1 | 29.86 | 2.26 | 15.89 | 7.9 | 1.64 | Ca8.7Mg1.1(PO4)6(OH)2 | |
| 7.5:1 | 33.96 | 2.17 | 17.27 | 9.56 | 1.67 | Ca9.1Mg0.9(PO4)6(OH)2 | |
| HA | 38.1 | — | 17.65 | — | 1.67 | Ca10(PO4)6(OH)2 | |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: