 图1
Fe3O4@MoO3@mSiO2纳米载体的合成及微波控制给药过程示意图
Figure1.
Schematic illustration showing the formation process of the Fe3O4@MoO3@mSiO2 nanocarriers and the subsequent drug loading and release control with microwave
图1
Fe3O4@MoO3@mSiO2纳米载体的合成及微波控制给药过程示意图
Figure1.
Schematic illustration showing the formation process of the Fe3O4@MoO3@mSiO2 nanocarriers and the subsequent drug loading and release control with microwave
引用本文:
唐冠鑫, 彭红霞, 胡传跃, 彭秧锡. 核-隔层-壳结构Fe3O4@MoO3@mSiO2纳米载体的合成及其药物可控释放[J]. 无机化学学报,
2018, 34(2): 337-345.
doi:
10.11862/CJIC.2018.041

Citation: TANG Guan-Xin, PENG Hong-Xia, HU Chuan-Yue, PENG Yang-Xi. Core-Spacer-Shell Structured Fe3O4@MoO3@mSiO2 Nanocarrier: Preparation and Controlled Release Drug[J]. Chinese Journal of Inorganic Chemistry, 2018, 34(2): 337-345. doi: 10.11862/CJIC.2018.041
Citation: TANG Guan-Xin, PENG Hong-Xia, HU Chuan-Yue, PENG Yang-Xi. Core-Spacer-Shell Structured Fe3O4@MoO3@mSiO2 Nanocarrier: Preparation and Controlled Release Drug[J]. Chinese Journal of Inorganic Chemistry, 2018, 34(2): 337-345. doi: 10.11862/CJIC.2018.041
核-隔层-壳结构Fe3O4@MoO3@mSiO2纳米载体的合成及其药物可控释放
摘要:
利用加热均匀、迅速、热平稳性好和安全性高的微波热响应来实现药物的微波可控释放。引入具有微波热响应性质、热稳定性和化学稳定性好的MoO3作为微波吸收物质,制备了核-隔层-壳结构Fe3O4@MoO3@mSiO2纳米药物载体。研究该纳米载体对药物布洛芬(IBU)的负载和微波响应可控释放过程。该纳米载体具有高的比表面积(222 cm2·g-1)和较大的孔隙体积(0.14 cm3· g-1)可用来负载药物。同时还具有较好的磁响应性,可实现药物的靶向给药,具有相对好的微波热响应性,可通过MoO3中间层吸收微波辐射实现药物的可控释放。结果表明,在持续微波辐射360 min时IBU的释放率达到86%,远远高于仅搅拌时的释放率。
-
关键词:
- Fe3O4@MoO3@mSiO2
- / 磁性
- / 吸波热转换性
- / 载药
- / 可控释放
English
Core-Spacer-Shell Structured Fe3O4@MoO3@mSiO2 Nanocarrier: Preparation and Controlled Release Drug
Abstract:
MoO3 interlayered Fe3O4@MoO3@mSiO2 core-spacer-shell structured drug nanocarrier was constructed to investigate loading and controllable release properties of ibuprofen (IBU). They possess high surface area of 222 cm2·g-1, provide large accessible pore volume of 0.14 cm3·g-1 for adsorption of drug molecules. At the same time they also have good magnetic responsiveness for drug targeting under foreigen magnetic under foreign magnetic fields, relatively good microwave heating conversion behavior for controlled release by microwave-triggered which is caused by MoO3 interlayer. The IBU release of over 86% under microwave discontinuous irradiation within 360 min outclasses the only stirring release.
-
Key words:
- Fe3O4@MoO3@mSiO2
- / magnetic
- / microwave heating conversion behavior
- / loading drug
- / controlled release
-
0 引言
以Fe3O4为核的“核-壳”结构纳米载体在靶向可控给药方面的研究领域已经成为国际热点问题之一[1-2]。具有靶向可控释放药物特性的纳米载体可以克服药物溶解性差、稳定性不好、代谢迅速等传统用药存在的问题,特别是在解决药物缺乏靶向选择性方面取得很好的效果[3-4]。而在药物载体的研究中提高药物的有效装载效率和可控释放药物是限制其应用的一个瓶颈。为了提高载体的药物负载量,研究者们通过对纳米载体进行表面修饰或者包覆介孔层来提高药物的负载量[5-6]。介孔二氧化硅纳米颗粒由于其较高的比表面积、可调节的孔径大小、低毒性、可生物降解性等,近年来在药物输送方面已成为最具有潜在应用前景的载体之一[7]。例如,Qiu等合成了二氧化硅包覆Fe3O4@ZnO的多功能纳米粒子并研究了其作为生物载体的性能[8]。纳米载体的可控给药的释放方式主要有内源性控释和外源性控释。内源性控释体系依赖于癌症部位特定的物理化学性质,如:高浓度的某些酶类,氧化还原环境及pH值的影响等[9]。然而,因为生物体内环境变化细微且难以控制,依靠生物体内部生理环境的细微改变来控制药的靶向和释放还存在很多大的挑战,相比之下体外刺激控制药物的靶向和释放研究得到了更多的关注[10]。在外源性控释方式(如磁场,红外福射,超声波福射和微波辐射等)中,由于微波对生物体的伤害程度小,在生物体内加热效率高,生物穿透性好,使得利用微波照射控制药物释放成为一种很有潜力的外部触发控药方式[11]。可以通过微波理疗仪控制一些参数如频率,功率密度,作用时间等在病变部位准确的局部加热,实现对靶向药物的控制释放。研究发现,氧化钼(MoO3)纳米材料具有优异的微波热响应特性,它们可以吸收一定频率的微波将其转化为热能,产生的热能可以加热纳米载体进而刺激控制药物释放[12-13]。并且,物质的微波吸收性能的大小与其禁带宽度有一定的关系,禁带宽度越小,其微波吸收性能越好。MoO3的禁带宽度是3.15 eV,小于ZnO(3.37 eV)和TiO2(3.2 eV)的禁带宽度。另一方面,MoO3具有很好的化学稳定性和热稳定性[14]。因此,MoO3纳米材料相比较于ZnO和TiO2来说具有更好的微波热响应性。也就是说,MoO3可吸收微波并使之转化为热能[15]。所产生的热量进一步加热纳米载体从而刺激药物的释放。
因此,本文制备了一种具有磁性核-隔层-壳结构的Fe3O4@MoO3@mSiO2药物载体。该载体的介孔二氧化硅壳层为载药层来提高药物的负载率。把磁性Fe3O4和具有微波吸收性能的MoO3纳米颗粒结合,同时赋予磁靶向性和微波可控释放的性能。为了研究该载体的药物可控释放性,以布洛芬为模板药物,并通过微波辐射响应来研究药物的可控释放。该纳米载体将会在生物医疗中的体内靶向给药和利用微波刺激进行可控释放药物方面具有潜在的应用前景。该纳米载体的合成及微波控制给药过程如图 1所示。
图1
Fe3O4@MoO3@mSiO2纳米载体的合成及微波控制给药过程示意图
Figure1.
Schematic illustration showing the formation process of the Fe3O4@MoO3@mSiO2 nanocarriers and the subsequent drug loading and release control with microwave
1 实验部分
1.1 主要试剂
实验药品均为分析纯,无需进一步纯化。氨水、浓硝酸、氢氧化钠购于衡阳市凯信化工试剂有限公司;十六烷基三甲基溴化铵(CTAB)购于湖南湘中化学试剂有限公司;三氯化铁(FeCl3·6H2O,99.0%)、钼酸铵((NH4)2MoO4,99.0%)、乙酸(CH3COOH,99.0%)、聚乙二醇(分子量6 000)购于天津科密欧化学试剂有限公司;实验用水为去离子水。
1.2 实验方法
磁性Fe3O4纳米颗粒的制备:称取1.4 g六水氯化铁溶于乙二醇,搅拌溶解后,继续将无水醋酸钠2.7 g、聚乙二醇1.0 g(分子量6 000)、磁力搅拌0.5 h后转入50 mL反应釜密封置于恒温箱中加热到200 ℃并恒温反应8 h。待反应体系温度冷却到室温后,将反应得到的黑色粉末悬液用磁铁分离。再通过去离子水和乙醇各洗涤3次,50 ℃下,干燥2 h,最终得到黑色的Fe3O4纳米粒子。
Fe3O4@MoO3纳米颗粒的制备:称取3.34 g的钼酸铵固体,溶于70 mL的蒸馏水,得到无色透明的溶液,然后,加入0.5 g Fe3O4和1 g CTAB,超声30 min,然后用磁力搅拌器60 ℃搅拌30 min,最后缓慢滴加乙酸溶液,滴加至pH为3.5。滴加完毕后,静置一段时间,即有前驱体黄色沉淀生成,用去离子水,无水乙醇分别洗涤3次,并过滤,然后在干燥箱中150 ℃烘干2 h,在空气中于500 ℃下煅烧2 h。
Fe3O4@MoO3@mSiO2纳米载体的制备:取0.05 g Fe3O4@MoO3加入到30 mL溶有0.15 g CTAB的乙醇溶液中,搅拌20 min后,加入0.4 mL正硅酸乙酯,搅拌60 min,然后边搅拌边缓缓加入2 mL去离子水,继续搅拌180 min,利用磁分离收集,所得到样品用乙醇和去离子水分别清洗3次,在80 ℃下干燥。最后500 ℃煅烧3 h,去除模板剂而得到Fe3O4 @MoO3@mSiO2。
纳米载体的载药过程:将0.1 g制备的Fe3O4 @MoO3、Fe3O4@SiO2和Fe3O4@MoO3@mSiO2纳米载体分别分散到50 mL布洛芬(IBU)的生理盐水(9 g·L-1)溶液中(0.3 g·L-1),然后磁力搅拌24 h。在药物负载过程中,分别在反应0、30、60、120、180、240、360和480 min时取上清液,通过紫外分光光度计测量溶液中IBU的浓度(石英比色皿)。通过朗伯-比尔定律计算纳米载体的载药量。
其中,M负是负载到纳米载体表面的药物IBU的质量,M初是药物负载过程中药物IBU的初始量。
药物释放过程:药物IBU的释放实验是在生理盐水中进行,为了研究微波辐射对药物释放率的影响,我们固定溶液的条件为37 ℃和pH=7.0,所采用的微波照射频率为2.45 GHz,具体实验过程是:称取0.1 g纳米颗粒,使其均匀分散在50 mL生理盐水中,在微波频率下辐射15 s取上清液测试,然后关掉微波发生器,持续搅拌30 min取上清液测试,然后再打开微波发生器进行微波辐射15 s,如此反复循环7次,每个循环中取的样品用紫外可见光光谱进行IBU浓度的测试,再进一步计算药物的释放率。
1.3 样品表征
德国Bruker D8 FOCUS多晶粉末X射线衍射(XRD)仪进行分析,(以Cu Kα1为X射线源,λ=0.154 056 nm,衍射角2θ为10°~80°,步长为10°·min-1,扫描速率为0.1°·s-1)。采用荷兰PEI公司的PHIIJIPS XL30 ESEMFEG环境扫描电镜(FESEM,200 kV)和日本JEOL公司的JEM-2010型透射电镜观察粒子的形貌和尺寸。MCR-3微波化学反应器观察载体的微波热转化效率。采用德国Bruker公司的tensor-27型红外光谱仪对样品进行红外光谱的测试。用表面吸附仪(TR2Star 3020)在液氮湿度下(77 K)测试样品的氮气吸附与解吸附过程。用日本岛津的UV-1800型紫外-可见分光光度计进行药物的负载和释放过程的紫外-可见光谱的测试。用美国Quantum Design公司的MPMS-XL-7超导量子干涉磁测量系统(SQUID)对样品的磁学性能进行测试。
2 结果与讨论
2.1 X射线衍射(XRD)分析
样品的相组成和结晶度由XRD进行测试分析。图 2是纯的Fe3O4颗粒、Fe3O4@MoO3颗粒以及Fe3O4 @MoO3@mSiO2纳米颗粒的XRD图。由图 2a可见,磁性核Fe3O4为尖晶石结构(PDF No.65-3107)。通过图 2b可以很清楚地检索到样品中除存在尖晶石结构的Fe3O4晶体外,22.9°,25.2°,26.4°,34.0°,37.5°,65.9°处的特征峰位与PDF标准卡片(PDF No.47-1081)吻合良好,对应结构为正交相的MoO3纳米晶体。并且,没有检测出其他杂峰,表明磁性Fe3O4核与MoO3壳层之间并未发生明显的化学反应。同时结合后面的TEM分析和EDS能谱分析,包覆了介孔SiO2后(图 2c)在17.2°左右出现了一个略凸起的宽峰,这就证明在该纳米颗粒中存在无定型的SiO2(用*表示)。
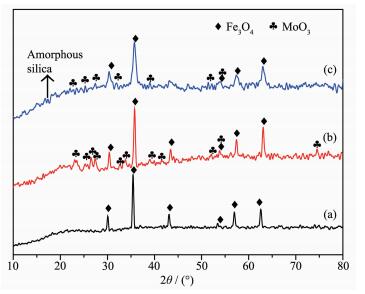 图2
Fe3O4 (a), Fe3O4@MoO3 (b)和Fe3O4@MoO3@mSiO2
(c)的XRD图
Figure2.
XRD patterns of the samples: Fe3O4 (a), Fe3O4@MoO3 (b) and Fe3O4@MoO3@mSiO2 (c)
图2
Fe3O4 (a), Fe3O4@MoO3 (b)和Fe3O4@MoO3@mSiO2
(c)的XRD图
Figure2.
XRD patterns of the samples: Fe3O4 (a), Fe3O4@MoO3 (b) and Fe3O4@MoO3@mSiO2 (c)
2.2 TEM和EDS分析
图 3为Fe3O4,Fe3O4@MoO3和Fe3O4@MoO3@ mSiO2的SEM图和TEM图以及与之相对应的EDS能谱图。图 3a为Fe3O4低倍率SEM电镜图,从图可以观测到制得的Fe3O4纳米粒子呈现球形单分散性,粒径分布均一且平均粒径大约为160 nm,其表面粗糙不平,结合XRD和电镜图显示的结果证明制备的Fe3O4纳米粒子是由小晶粒堆积而成。Fe3O4纳米颗粒的EDS能谱图(图 3g)进一步证实了在Fe3O4样品中同时存在O和Fe元素。图 3b为MoO3包覆的Fe3O4纳米粒子的SEM电镜图,如图所示Fe3O4@MoO3纳米颗粒依然保持球形和较好的分散性,粒径大约为200 nm,包覆MoO3后粒径有些增大。Fe3O4@MoO3纳米颗粒的EDS能谱图(图 3h)进一步证实了在Fe3O4@MoO3样品中同时存在Mo,O和Fe元素。进一步包覆介孔SiO2的SEM图如图 3(c)所示,由电镜图进行形貌和粒径分析发现包覆介孔SiO2后的纳米粒子表面相对于Fe3O4和Fe3O4@MoO3纳米颗粒显得光滑,看不到小颗粒状结构,粒径约为240 nm,同样其粒径分布也呈现出均匀分散的球形形貌。Fe3O4@MoO3@mSiO2纳米颗粒的EDS能谱图(图 3h)进一步证实在Fe3O4@MoO3@mSiO2样品中同时存在Si,Mo,O和Fe元素。图 3(d~f)给出的是样品Fe3O4,Fe3O4@MoO3和Fe3O4@MoO3@mSiO2的TEM图。从TEM照片可看出,3种样品均具有很好的分散性。对样品Fe3O4@MoO3的TEM图(图 3e)进行观察发现,在Fe3O4纳米颗粒的表面包覆了一层MoO3,并由单个的MoO3纳米颗粒组成的。而且,Fe3O4@MoO3纳米颗粒的表面比纯的MoO3的表面要粗糙很多,这也证明了在Fe3O4表面包覆上了一层MoO3层。图 3f是Fe3O4@MoO3@mSiO2纳米颗粒的TEM照片。由纳米颗粒的内部和边缘明显的衬度对比可看出制备的Fe3O4@MoO3@mSiO2纳米颗粒具有良好的“核-隔层-壳”结构。核部的磁性四氧化铁为黑色的球形颗粒,其平均粒径大约为200 nm。图 3f附图中看到包覆在Fe3O4磁性核外部的MoO3纳米颗粒。同时,均匀包覆的二氧化硅层显示的是灰色,其厚度大约为40 nm。值得注意的是,在Fe3O4 @MoO3@mSiO2纳米颗粒的表面能够清楚的看到介孔二氧化硅的孔道。综上分析,证明我们已经成功的制备出了具有“核-隔层-壳”结构的球形Fe3O4@MoO3@mSiO2纳米载体。
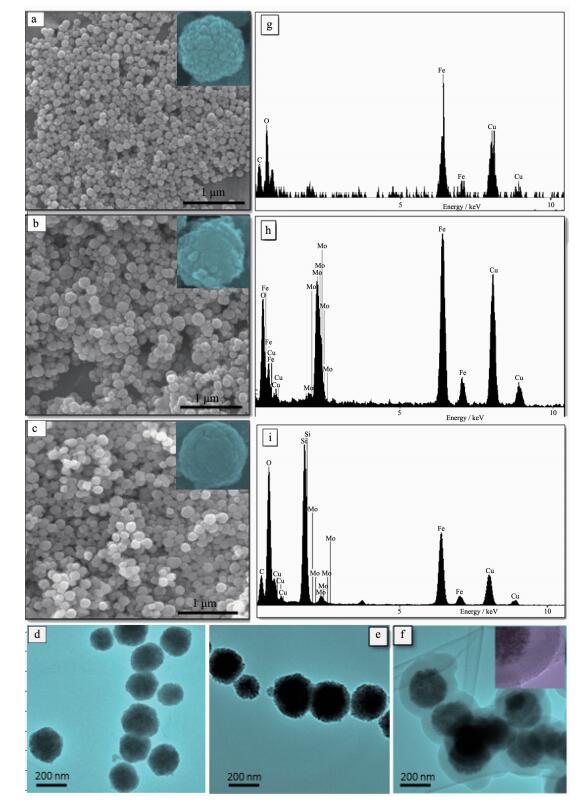 图3
Fe3O4 (a, g), Fe3O4@MoO3 (b, h), Fe3O4@MoO3@mSiO2 (c, i)的SEM图与EDS能谱图; Fe3O4 (d), Fe3O4@MoO3 (e), Fe3O4@MoO3@mSiO2 (f)的TEM图
Figure3.
SEM, TEM images and EDS spectrum of Fe3O4 (a, d, g), Fe3O4@MoO3 (b, e, h), Fe3O4@MoO3@mSiO2 (c, f, i)
图3
Fe3O4 (a, g), Fe3O4@MoO3 (b, h), Fe3O4@MoO3@mSiO2 (c, i)的SEM图与EDS能谱图; Fe3O4 (d), Fe3O4@MoO3 (e), Fe3O4@MoO3@mSiO2 (f)的TEM图
Figure3.
SEM, TEM images and EDS spectrum of Fe3O4 (a, d, g), Fe3O4@MoO3 (b, e, h), Fe3O4@MoO3@mSiO2 (c, f, i)
2.3 介孔结构分析
为了研究Fe3O4@MoO3@mSiO2纳米颗粒的介孔性能,我们进行了液氮吸附-脱附测试(见图 4)。根据国际理论和应用化学联合会的标准,样品的吸附-脱附曲线表现出典型的有H4型滞留环的Ⅳ型等温曲线,该曲线一般与介孔二氧化硅的孔道类型有关。计算得到Fe3O4@MoO3@mSiO2纳米颗粒的BET比表面积和孔容积分别是222 cm2·g-1和0.14 cm3·g-1。从图 4插图孔径分布曲线中我们也进一步证实Fe3O4@MoO3@mSiO2纳米颗粒的介孔孔径为2.40 nm左右。液氮吸附-解吸附结果显示纳米颗粒的孔道为介孔结构,因此该纳米颗粒适合作为药物载体来运输药物。综上所述,介孔二氧化硅成功包覆在纳米Fe3O4@MoO3颗粒表面。
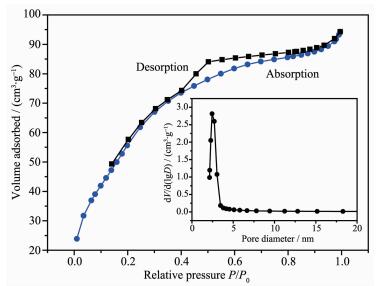 图4
Fe3O4@MoO3@mSiO2纳米颗粒的液氮吸附-脱附图, 插图为介孔的孔径分布
Figure4.
Pore diameter distribution and N2 adsorption-desorption isotherms of Fe3O4@MoO3@mSiO2
图4
Fe3O4@MoO3@mSiO2纳米颗粒的液氮吸附-脱附图, 插图为介孔的孔径分布
Figure4.
Pore diameter distribution and N2 adsorption-desorption isotherms of Fe3O4@MoO3@mSiO2
2.4 红外光谱(FTIR)分析
为了研究纳米颗粒的结构及其是否成功地负载上药物IBU,进行了Fe3O4@MoO3@mSiO2、Fe3O4@ MoO3@mSiO2-IBU和IBU的红外光谱研究。图 5a是Fe3O4@MoO3@mSiO2的红外光谱图。谱图中1 632 cm-1是H2O的吸收峰,这就说明在纳米颗粒的表面存在大量的-OH和H2O分子,这就可以通过羟基与药物IBU分子进行连接从而负载上大量的药物分子。同时,在光谱图中还存在Si-O-Si(1 083 cm-1),Si-OH(948 cm-1),和Fe-O(574 cm-1)[16]。装载IBU后的纳米颗粒Fe3O4@MoO3@mSiO2-IBU和纯的IBU的红外光谱图如图 7(b, c)所示, 谱图 7b中位于1 483 cm-1处的特征吸收峰对应于IBU药物分子中的C=C伸缩振动吸收峰,而在1 756 cm-1处的特征吸收峰对应于IBU药物分子中的C=O伸缩振动吸收峰,位于2 924 cm-1处的特征吸收峰对应于IBU药物分子中CH2吸收峰[17]。装载后的Fe3O4@MoO3@mSiO2出现了IBU药物分子的特征吸收峰,说明其成功载药。
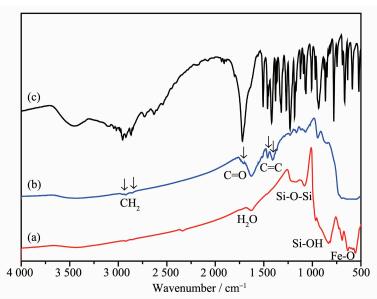 图5
Fe3O4@MoO3@mSiO2 (a), Fe3O4@MoO3@mSiO2-IBU (b)和IBU (c)的红外光谱图
Figure5.
FT-IR Spectra of Fe3O4@MoO3@mSiO2 (a), Fe3O4@ MoO3@mSiO2-IBU (b) and IBU (c)
图5
Fe3O4@MoO3@mSiO2 (a), Fe3O4@MoO3@mSiO2-IBU (b)和IBU (c)的红外光谱图
Figure5.
FT-IR Spectra of Fe3O4@MoO3@mSiO2 (a), Fe3O4@ MoO3@mSiO2-IBU (b) and IBU (c)
2.5 磁性与微波热转化性能
图 6为Fe3O4@MoO3@mSiO2-IBU的外加磁场下的宏观磁性表征照片。由图可见均匀分散在溶液中的纳米颗粒在外加磁场的作用下快速聚集到磁铁附近,结果说明该纳米颗粒具有快速响应外部磁场的性能和分散性能,可以用在磁性分离和磁靶向药物载体等方面。
 图6
Fe3O4@MoO3@mSiO2-IBU的磁响应图
Figure6.
Magnetic response of Fe3O4@MoO3@mSiO2-IBU
图6
Fe3O4@MoO3@mSiO2-IBU的磁响应图
Figure6.
Magnetic response of Fe3O4@MoO3@mSiO2-IBU
为了验证微波能否用来刺激控制药物释放,进行了Fe3O4@MoO3@mSiO2纳米颗粒吸收微波后的微波热响应测试。该测试是在室温条件下进行的,测试的微波频率为2.45 GHz,这与生物医疗上所用到的频率范围一致。生理盐水、Fe3O4、Fe3O4@MoO3和Fe3O4@MoO3@mSiO2纳米颗粒在微波辐射下的时间-温度曲线如图 7所示。从图中可看出,4种物质表现出了不同的微波热响应性能。其微波热响应速率的大小为:生理盐水 < Fe3O4 < Fe3O4@MoO3@mSiO2 < Fe3O4@MoO3。因此,Fe3O4@MoO3@mSiO2纳米颗粒具有很好的微波热效应,在60 s内就可以快速升温到56 ℃。但是生理盐水和Fe3O4溶液其微波热效应相对要差一些(60 s后分别升温到38和49 ℃)。实验测试结果显示Fe3O4@MoO3@mSiO2和Fe3O4@MoO3纳米颗粒均具有很好的微波吸收性能和微波热转换性能。作为二者之间共有的物质,MoO3具有很好的微波吸收性能,在相同的微波辐射频率下,MoO3可以快速地将电磁能转换为热能。这是因为MoO3具有压电性能,可以将电磁波发射到振动能量的表面而转换成热能。Fe3O4@MoO3@mSiO2纳米颗粒与Fe3O4 @MoO3相比,其微波热转换性能有所降低,这可能是由于MoO3的浓度相对减少了(介孔二氧化硅的存在降低了MoO3纳米颗粒的浓度)。但是,Fe3O4@ MoO3@mSiO2纳米颗粒仍具有比较明显的微波热响应性能,适合于通过局部加热进行药物的可控释放和治疗癌症的目的。
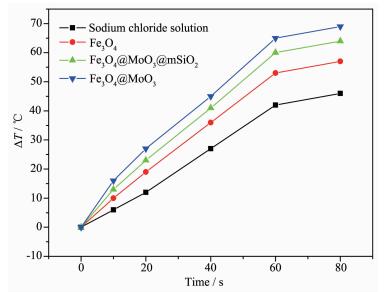 图7
生理盐水、Fe3O4、Fe3O4@MoO3和Fe3O4@MoO3@ mSiO2在2.45 GHz时的微波热响应图
Figure7.
Heating curves of sodium chloride solution, Fe3O4, Fe3O4@MoO3 and Fe3O4@MoO3@mSiO2 under microwave irradiation at 2.45 GHz
图7
生理盐水、Fe3O4、Fe3O4@MoO3和Fe3O4@MoO3@ mSiO2在2.45 GHz时的微波热响应图
Figure7.
Heating curves of sodium chloride solution, Fe3O4, Fe3O4@MoO3 and Fe3O4@MoO3@mSiO2 under microwave irradiation at 2.45 GHz
2.6 药物装载和释放性能
本文选择药物IBU作为模型药物研究药物的负载和释放性能。为了避免其他因素对实验结果的影响,在37 ℃时进行药物的负载和释放实验,实验在生理盐水中进行(9 g·L-1,模拟人体体液)。
Fe3O4@MoO3@mSiO2纳米颗粒的介孔层为药物分子的负载提供了较大的孔体积,其平均孔径大小为2.40 nm,是IBU药物分子直径(0.60 nm)的4倍[18]。IBU分子与介孔二氧化硅表面上的羟基之间可以氢键相连或物理吸附,从而使得IBU分子装载到介孔二氧化硅层的孔内,然后通过微波加热促使二者之间的氢键断裂或作用力破坏,使得IBU药物分子从载体上释放出来,从而实现微波可控释放的目的。
图 8a、8b和8c给出了室温下在生理盐水中,Fe3O4@SiO2、Fe3O4@MoO3和Fe3O4@MoO3@mSiO2纳米颗粒药物的负载过程和药物负载了不同时间后溶液的紫外吸收光谱曲线。在开始的时候药物的负载率迅速增大,在30 min内其负载率均达到约40%,但是随后负载率增长缓慢。由图可以看出IBU在225 nm处有最大吸收峰,并且随着时间的延长,所对应的吸光强度值减小,这就说明溶液中药物的浓度是随负载时间的延长而减小的。480 min后Fe3O4@SiO2和Fe3O4@MoO3的药物负载率分别大约为44%和43%,而大约有70%的IBU分子装载到了Fe3O4@MoO3@mSiO2纳米颗粒的孔内或表面。相比于Fe3O4@SiO2和Fe3O4@MoO3, Fe3O4@MoO3@mSiO2纳米颗粒具有较高的装载率,归功于介孔二氧化硅的多孔表面和其强大的装载能力。
 图8
纳米颗粒的药物负载动力学分析: UV-Vis光谱图和药物负载率与时间的函数图(插图)
Figure8.
Kinetic analysis of drug loading: UV-Vis spectrophotometer and drug loading versus time (inset)
图8
纳米颗粒的药物负载动力学分析: UV-Vis光谱图和药物负载率与时间的函数图(插图)
Figure8.
Kinetic analysis of drug loading: UV-Vis spectrophotometer and drug loading versus time (inset)
为了进一步确定微波照射对载体释放药物的影响,我们对比研究了微波照射和无微波仅搅拌药物释放速率的变化曲线,如图 9所示。图中微波照射和无微波仅搅拌情况下,载体上药物释放速率的变化十分明显,说明该纳米载体具有很好的刺激响应释放性能。从Fe3O4@MoO3@mSiO2-IBU在微波照射和无微波仅搅拌情况下的药物释放速率变化图可见,在没有微波照射下,纳米载体对IBU药物分子的释放速率非常慢,搅拌下经过90 s,IBU药物分子的释放量仅为1.5%,然而,当把药物释放体系转到微波照射的环境时IBU药物分子的释放速率有一个明显的提升,微波照射90 s药物分子的释放量达到了82%。为了研究MoO3隔层对纳米载体微波响应释放性能的影响,我们以相同的方法研究了没有包覆MoO3隔层的纳米载体Fe3O4@SiO2-IBU的药物释放性能,如图 9所示。Fe3O4@SiO2-IBU在没有微波照射下IBU药物分子的释放速率也很慢,90 s内释放量仅为1.5%,微波照射下释放速率也有很大的提升,微波照射90 s药物的总释放量达到了15%。结果说明没有包覆MoO3隔层的纳米载体也具有一定的微波热响应性,因为Fe3O4本身也具有一定的微波吸收性能,但是包覆有MoO3隔层的纳米载体具有更好的微波吸收性能。
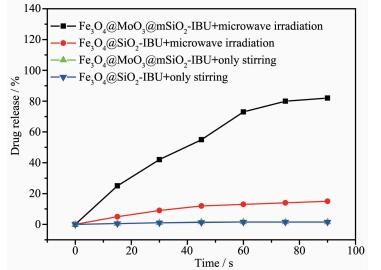 图9
Fe3O4@MoO3@mSiO2-IBU和Fe3O4@SiO2-IBU在微波照射和没有微波照射仅搅拌下IBU的对比释放图
Figure9.
Release profiles of IBU from nanocarriers of Fe3O4
@MoO3@mSiO2-IBU and Fe3O4@SiO2-IBU with or without microwave irradiation
图9
Fe3O4@MoO3@mSiO2-IBU和Fe3O4@SiO2-IBU在微波照射和没有微波照射仅搅拌下IBU的对比释放图
Figure9.
Release profiles of IBU from nanocarriers of Fe3O4
@MoO3@mSiO2-IBU and Fe3O4@SiO2-IBU with or without microwave irradiation
微波对比释放研究表明微波照射能大大提高纳米载体的药物释放速率,包覆有MoO3隔层的纳米颗粒具有很好的微波热响应性,在微波照射下能迅速吸收微波产生热量,破坏载体与药物分子之间的作用力,致使装载在纳米载体介孔中的药物分子释放出来,表现为Fe3O4@MoO3@mSiO2-IBU纳米载体对微波热响应,促进药物释放性能。
Fe3O4@MoO3@mSiO2-IBU体系的微波控释过程用紫外-可见光谱实时监测。为了研究其可控性和重复性,实验过程采用的是先用微波辐射实验体系监测其药物释放,然后停止微波辐射,只采用搅拌促使药物释放,以这样的形式作为一个循环。在每个循环中,先对样品用微波发生器辐射15 s,然后将微波发生器关掉,仅在37 ℃时搅拌30 min。图 10的结果显示,IBU药物分子在有微波辐射时被释放出来,然后,当关掉微波发生器后就抑制了药物IBU分子的释放。在实验初始阶段,对Fe3O4@MoO3@mSiO2-IBU溶液进行首次辐射了15 s后,大约有26%的IBU分子被释放出来,但是,当停止微波辐射仅进行30 min搅拌,只有大约2%的IBU分子被释放出来,远远小于微波辐射时的药物释放率。结果表明,负载了IBU分子的纳米颗粒在微波照射下会快速的释放出IBU分子,证明微波在药物的释放过程中起到了促进药物释放的作用。并且,负载了药物之后的纳米颗粒可以通过调节微波福射的开/关状态来精确的控制药物的释放。随着体系药物释放时间的延长,药物的释放曲线趋于平坦,经过了7个循环后药物IBU分子的释放率约为86%。但是,在该释放过程中,可通过调节微波辐射的时间和有无微波辐射实现药物释放的精确控制。这有可能是因为微波热效应与微波辐射时间有关系,随着辐射时间的延长微波热效应增强,同时样品的温度也会升高。因此,药物IBU分子的累积释放率随着微波辐射时间的延长而增大。这就表明,可以通过微波照射实现药物的可控释放,而且以MoO3为微波吸收层时其微波可控释放效果优于以ZnO和TiO2为微波吸收层时的效果。所以,所制备的Fe3O4@MoO3@mSiO2纳米载体通过调节微波辐射的状态实现药物的可控释放是可行的。
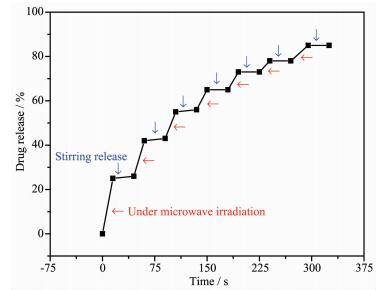 图10
微波照射和搅拌下Fe3O4@MoO3@mSiO2-IBU对药物IBU的可控释放行为
Figure10.
Cumulative release profiles of IBU from Fe3O4@ MoO3@mSiO2-IBU upon 2.45 GHz microwave irradiation
图10
微波照射和搅拌下Fe3O4@MoO3@mSiO2-IBU对药物IBU的可控释放行为
Figure10.
Cumulative release profiles of IBU from Fe3O4@ MoO3@mSiO2-IBU upon 2.45 GHz microwave irradiation
3 结论
本文制备了一种以Fe3O4为“核”,MoO3作为中间微波响应层,介孔二氧化硅为“壳”的Fe3O4@MoO3 @mSiO2纳米载药颗粒,并研究了其对药物布洛芬(IBU)的负载和可控释放行为。所制备的纳米载体为球形结构,具有较好的磁响性,可以实现药物的靶向给药。有较大的比表面积、孔体积和较大的孔径,可实现药物的有效负载;同时具有较好的微波热转化性,可以通过MoO3中间层吸收微波辐射实现药物的可控释放。实验表明,在微波持续辐射经过7个循环后药物IBU的释放率达到86%,在生物医疗中有很好的应用前景。
-
-
[1]
Zhu Y F, Fang Y, Kaskel S. J. Phys. Chem. C, 2010, 114:16382-16388
-
[2]
Cui X J, Mathe D, Kovács N, et al. Bioconjugate Chem., 2016, 27(2):319-328 doi: 10.1021/acs.bioconjchem.5b00338
-
[3]
Fu G L, Sanjay S T, Dou M W, et al. Nanoscale, 2016, 8(10):5422-5427 doi: 10.1039/C5NR09051B
-
[4]
Cheng L, Wang C, Ma X X, et al. Adv. Funct. Mater., 2013, 23:272-280
-
[5]
Luo Z, Cai K, Hu Y. Adv. Mater., 2012, 24:431-435
-
[6]
Wong T W, Chan L W, Kho S B, et al. J. Controlled Release, 2002, 84:99-114 doi: 10.1016/S0168-3659(02)00237-7
-
[7]
Zhao W W, Cui B, Peng H X, et al. J. Phys. Chem. C, 2015, 119(8):4379-4386
-
[8]
Qiu H J, Cui B, Li G M, et al. J. Phys. Chem. C, 2014, 118:14929-14937
-
[9]
Peng H X, Cui B, Wang Y S. Mater. Sci. Eng., C, 2015, 46:253-263 doi: 10.1016/j.msec.2014.10.022
-
[10]
Peng H X, Cui B, Zhao W W, et al. Expert Opin. Drug Delivery, 2015, 12(9):1397-1409
-
[11]
Qiu H J, Cui B, Zhao W W, et al. J. Mater. Chem. B, 2015, 3:6919-6927
-
[12]
Peng H X, Cui B, Wang Y S. New J. Chem., 2015, 40:1460-1466
-
[13]
Peng H X, Wang X, Hu C Y, et al. New J. Chem., 2016, 40:7911-7920 doi: 10.1039/C6NJ01651K
-
[14]
Bao T, Yin W, Zheng X, et al. Biomaterials, 2016, 76:11-18
-
[15]
Su H, Zhang H W, Tang X L, et al. Mater. Chem. Phys., 2007, 102:271-274
-
[16]
Wang Y S, Peng H X, Cui B. Mater. Chem. Phys., 2014, 146:330-338
-
[17]
Zhang X, Yang P P, Dai Y L, et al. Adv Funct. Mater., 2013, 23:4067-4078
-
[18]
彭红霞, 胡传跃, 吴腾宴, 等.无机化学学报, 2016, 7:1154-1160 doi: 10.11862/CJIC.2016.159PENG Hong-Xia, HU Chuan-Yue, WU Teng-Yan, et al. Chinese J. Inorg. Chem., 2016, 7:1154-1160 doi: 10.11862/CJIC.2016.159
-
[1]
-

图 1 Fe3O4@MoO3@mSiO2纳米载体的合成及微波控制给药过程示意图
Figure 1 Schematic illustration showing the formation process of the Fe3O4@MoO3@mSiO2 nanocarriers and the subsequent drug loading and release control with microwave

图 2 Fe3O4 (a), Fe3O4@MoO3 (b)和Fe3O4@MoO3@mSiO2 (c)的XRD图
Figure 2 XRD patterns of the samples: Fe3O4 (a), Fe3O4@MoO3 (b) and Fe3O4@MoO3@mSiO2 (c)

图 3 Fe3O4 (a, g), Fe3O4@MoO3 (b, h), Fe3O4@MoO3@mSiO2 (c, i)的SEM图与EDS能谱图; Fe3O4 (d), Fe3O4@MoO3 (e), Fe3O4@MoO3@mSiO2 (f)的TEM图
Figure 3 SEM, TEM images and EDS spectrum of Fe3O4 (a, d, g), Fe3O4@MoO3 (b, e, h), Fe3O4@MoO3@mSiO2 (c, f, i)

图 4 Fe3O4@MoO3@mSiO2纳米颗粒的液氮吸附-脱附图, 插图为介孔的孔径分布
Figure 4 Pore diameter distribution and N2 adsorption-desorption isotherms of Fe3O4@MoO3@mSiO2

图 5 Fe3O4@MoO3@mSiO2 (a), Fe3O4@MoO3@mSiO2-IBU (b)和IBU (c)的红外光谱图
Figure 5 FT-IR Spectra of Fe3O4@MoO3@mSiO2 (a), Fe3O4@ MoO3@mSiO2-IBU (b) and IBU (c)

图 7 生理盐水、Fe3O4、Fe3O4@MoO3和Fe3O4@MoO3@ mSiO2在2.45 GHz时的微波热响应图
Figure 7 Heating curves of sodium chloride solution, Fe3O4, Fe3O4@MoO3 and Fe3O4@MoO3@mSiO2 under microwave irradiation at 2.45 GHz

图 8 纳米颗粒的药物负载动力学分析: UV-Vis光谱图和药物负载率与时间的函数图(插图)
Figure 8 Kinetic analysis of drug loading: UV-Vis spectrophotometer and drug loading versus time (inset)
(a) Fe3O4@SiO2, (b) Fe3O4@MoO3, (c) Fe3O4@MoO3@mSiO2

图 9 Fe3O4@MoO3@mSiO2-IBU和Fe3O4@SiO2-IBU在微波照射和没有微波照射仅搅拌下IBU的对比释放图
Figure 9 Release profiles of IBU from nanocarriers of Fe3O4 @MoO3@mSiO2-IBU and Fe3O4@SiO2-IBU with or without microwave irradiation
-


 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 5
- 文章访问数: 2441
- HTML全文浏览量: 434
 下载:
下载:
