 图1
(a) 三嗪基氮化碳结构; (b)三均三嗪基氮化碳结构; (c) melon基氮化碳结构
Figure1.
(a) Triazine-, (b) Tri-s-triazine-and (c) melon-based carbon nitride
图1
(a) 三嗪基氮化碳结构; (b)三均三嗪基氮化碳结构; (c) melon基氮化碳结构
Figure1.
(a) Triazine-, (b) Tri-s-triazine-and (c) melon-based carbon nitride
引用本文:
王辉, 张晓东, 谢毅. 聚合物氮化碳材料光激发过程研究进展[J]. 无机化学学报,
2017, 33(11): 1897-1913.
doi:
10.11862/CJIC.2017.249

Citation: WANG Hui, ZHANG Xiao-Dong, XIE Yi. Recent Progresses on the Photoexcitation Processes of Polymeric Carbon Nitride-Based Materials[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(11): 1897-1913. doi: 10.11862/CJIC.2017.249
Citation: WANG Hui, ZHANG Xiao-Dong, XIE Yi. Recent Progresses on the Photoexcitation Processes of Polymeric Carbon Nitride-Based Materials[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(11): 1897-1913. doi: 10.11862/CJIC.2017.249
聚合物氮化碳材料光激发过程研究进展
摘要:
近年来,不含金属元素的聚合物氮化碳半导体以其独特的材料组成和电子结构特征吸引了研究人员的广泛兴趣,在诸如光催化、光致发光、光电化学等光激发相关领域具有潜在的应用前景。聚合物氮化碳光激发过程由能带结构、载流子行为、激子效应等因素所主导,对于这一过程的研究在材料性能的优化、应用领域的拓展以及应用机制的理解等方面具有重要意义。围绕这一主题,本文综述了近年来聚合物氮化碳材料光激发过程研究中取得的最新进展,简单介绍了聚合物半导体光激发过程的研究方法,分别讨论了光激发下材料中的载流子行为和激子过程,进而总结了常用改性策略对于材料光激发过程的调控机制,此外还对材料光响应特性功能化的研究进行了概括。
English
Recent Progresses on the Photoexcitation Processes of Polymeric Carbon Nitride-Based Materials
Abstract:
Recently, the metal-free polymeric carbon nitride has drawn tremendous attention by virtue of its unique material composition and electronic structure, holding great promise in optical excitation related applications, such as photocatalysis, photoluminescence, and photoelectrochemistry. The investigation of photoexcitation processes dominated by energy band structure, charge carrier behaviors and excitonic effects in polymeric carbon nitride are of great significance in understanding mechanisms, optimizing performance and expanding application. Focusing on this topic, we review recent advances in research on the photoexcitation processes of polymeric carbon nitride-based materials, by beginning with a brief introduction of the characterization techniques in these works. We then discuss the charge carrier and exciton aspects in the photoexcitation processes. The principles of universal modification strategies on photoexcitation processes are summarized. Also, the applications of the photoexcitation processes in this polymeric semiconductor are highlighted.
-
Key words:
- polymeric carbon nitride
- / photoexcitation
- / charge carrier
- / excitonic effects
-
0 引言
作为一种重要的不含金属元素的半导体材料,聚合物氮化碳(有时也被称作石墨相氮化碳)以其广泛的原料来源、良好的结构稳定性、独特的电子结构等优势吸引了众多研究人员的目光,在光催化、光致发光、光电催化等领域受到了广泛关注[1-8]。鉴于上述研究与材料的光响应性质紧密相关,对于聚合物氮化碳材料光激发过程的研究具有非常重要的意义,不仅有助于从本质上对材料相关应用进行理解,同时也为进一步的材料优化设计提供理论指导。
聚合物氮化碳材料具有类石墨的层状结构,根据组成单元差异,可以分为三嗪基(图 1a)和三均三嗪基(图 1b)2种同素异形体,结构中碳、氮原子之间产生sp2杂化,形成具有高度离域π键的芳香共轭环状结构[4-9]。由于同素异形体中氮孔大小和氮原子化学环境的差异,三嗪基和三均三嗪基结构的具有不同的稳定性和光激发特性,而理论模拟显示具有三均三嗪结构单元的材料稳定性更好。另一方面,实验结果显示,在常压下由三聚氰胺、二氰二胺、尿素等前驱物热聚制备的聚合物氮化碳存在着聚合不完全的情况,此时结构主要是由三均三嗪单体连接形成的链(melon,图 1c)通过未聚合氨基产生的氢键相连接构成[10-14]。聚合物氮化碳的这种独特的结构特征,一方面给材料结构与应用性能之间构效关系的研究带来了难度,另一方面却也为材料的光激发过程调控提供了丰富多样的策略。
图1
(a) 三嗪基氮化碳结构; (b)三均三嗪基氮化碳结构; (c) melon基氮化碳结构
Figure1.
(a) Triazine-, (b) Tri-s-triazine-and (c) melon-based carbon nitride
目前,聚合物氮化碳材料光激发过程研究重点集中在基于能带理论的光生载流子过程上,诸如能带结构调整和载流子行为优化,都被认为是最有效的材料光激发过程调控策略[2-8]。除此之外,考虑到材料的结构特点,聚合物氮化碳较小的介电常数将会导致其具有较弱的屏蔽效应,使得体系中光生物种之间存在强的相互作用,而这种强相互作用将会对材料光激发过程及其应用产生重要影响[15]。最近,得益于材料合成方法与表征技术的发展,聚合物氮化碳光激发过程研究取得了丰富成果,为材料的应用机制研究以及性能优化提供了更深入的理解。本文将聚焦聚合物氮化碳材料,概述基于载流子和激子观点的光激发过程研究、调控策略以及应用拓展等方面的进展,并对该体系存在的主要问题及发展趋势进行总结与展望。
1 聚合物氮化碳光激发过程研究
1.1 聚合物半导体材料的光激发过程研究方法
考虑到材料的光吸收、能带结构、载流子迁移等与光激发过程紧密相关的特征和行为主要由其电子结构特征所决定,通过理论计算对材料电子结构进行模拟,进而预测材料光激发物种的行为,能够为进一步的实验研究提供指导。近年来,随着高性能计算技术及计算规模的快速发展,基于密度泛函理论的第一性原理计算逐渐成为一种研究多电子体系电子结构的通用方法,在聚合物半导体光激发过程研究方面具有重要应用[16-19]。
例如,Zhang等通过第一性原理计算研究了聚合物氮化碳块材和纳米片的电子结构(图 2),结果显示较之相应块材,纳米片中碳原子和氮原子之间的p轨道杂化增强,从而具有显著提升的导带边电荷密度分布,由此推测氮化碳纳米片具有较之块材更高的光相应性能[20];Ma等考察了不同非金属元素掺杂下聚合物氮化碳材料的HOMO和LUMO轨道的电荷分布,指出非金属元素掺杂能够有效促进光生载流子分离和提升材料电荷迁移率[21]。需要指出的是,聚合物材料实际结构的复杂性,使得理论模型并不能完全反映其结构特征,但总的来说,利用理论计算研究材料电子结构,进而对其光激发过程进行定性的判断依旧具有重要意义。
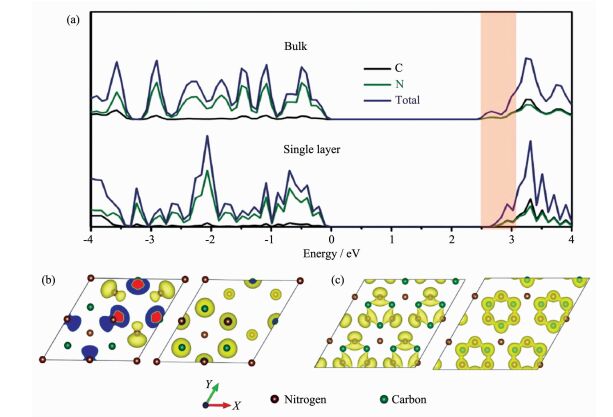 图2
聚合物氮化碳纳米片和块材的理论计算研究: (a)态密度, (b)块材的价带和导带电子波函数图,
(c)纳米片的价带和导带电子波函数图[20]
Figure2.
(a) Calculated density of states (DOS) of the single-layered and bulk polymeric carbon nitride, Kohn-Sham orbitals for the valence and conduction bands of (b) bulk and (c) single-layered polymeric carbon nitride, respectively[20]
图2
聚合物氮化碳纳米片和块材的理论计算研究: (a)态密度, (b)块材的价带和导带电子波函数图,
(c)纳米片的价带和导带电子波函数图[20]
Figure2.
(a) Calculated density of states (DOS) of the single-layered and bulk polymeric carbon nitride, Kohn-Sham orbitals for the valence and conduction bands of (b) bulk and (c) single-layered polymeric carbon nitride, respectively[20]
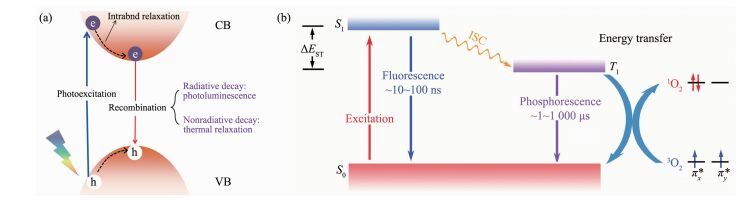
光致发光光谱技术是研究材料激发态辐射衰减过程最直接、有效的实验方法。通过稳态光谱分析,研究人员能够得到体系能带结构、缺陷能级、不同自旋态激子分布等信息, 而相应时间分辨发光光谱技术则能为材料光激发过程中光生物种的动力学研究提供有力支持[22-25]。近期,Wang等首次通过磷光光谱技术证实了聚合物氮化碳材料中存在大量的三线态激子,并结合荧光测试结果初步给出了体系激子态能级分布;他们进一步利用低温和瞬态光谱分析发现了材料的P型延迟荧光现象(图 3),不仅证实了体系中存在强烈的多体效应,同时也为材料光激发应用提供了新思路[26]。虽然光致发光谱技术能够直观的反应体系激发态的能级、衰减等信息,然而一旦考虑到体系中的飞秒尺度快速热振动辅助弛豫过程,潜在的非辐射衰减过程以及复杂的光学明、暗态布局,使得仅利用光致发光谱研究很难刻画出完整的光物理模型。
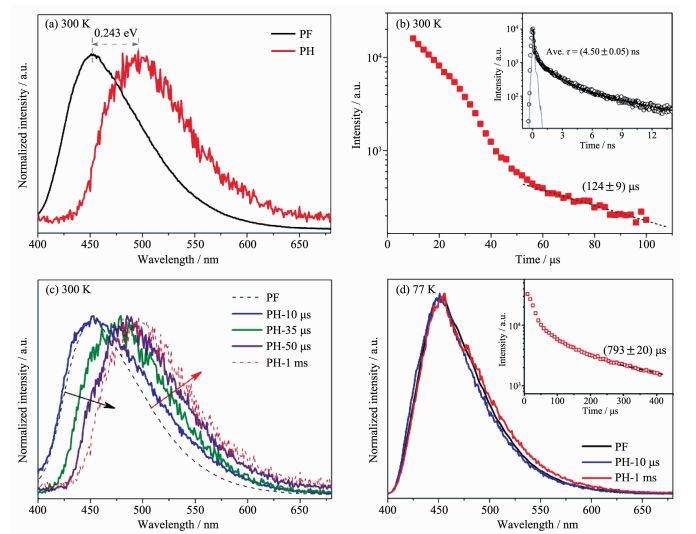 图3
聚合物氮化碳材料的光致发光光谱研究: 300 K下, (a)稳态荧光/磷光光谱, (b)时间分辨荧光/磷光光谱,
(c)不同延迟时间的磷光光谱, (d)低温下稳态荧光/磷光光谱及时间分辨磷光光谱[26]
Figure3.
(a) Normalized steady-state prompt fluorescence (PF) and phosphorescence (PH) spectra at 300 K, (b) Time-resolved PH kinetics at 300 K, Inset: time-resolved PF kinetics at 300 K, (c) Normalized steady-state PH spectra at different delay times at 300 K, (d) Normalized steady-state PF and PH spectra at 77 K, Inset: time-resolved PH kinetics at 77 K[26]
图3
聚合物氮化碳材料的光致发光光谱研究: 300 K下, (a)稳态荧光/磷光光谱, (b)时间分辨荧光/磷光光谱,
(c)不同延迟时间的磷光光谱, (d)低温下稳态荧光/磷光光谱及时间分辨磷光光谱[26]
Figure3.
(a) Normalized steady-state prompt fluorescence (PF) and phosphorescence (PH) spectra at 300 K, (b) Time-resolved PH kinetics at 300 K, Inset: time-resolved PF kinetics at 300 K, (c) Normalized steady-state PH spectra at different delay times at 300 K, (d) Normalized steady-state PF and PH spectra at 77 K, Inset: time-resolved PH kinetics at 77 K[26]
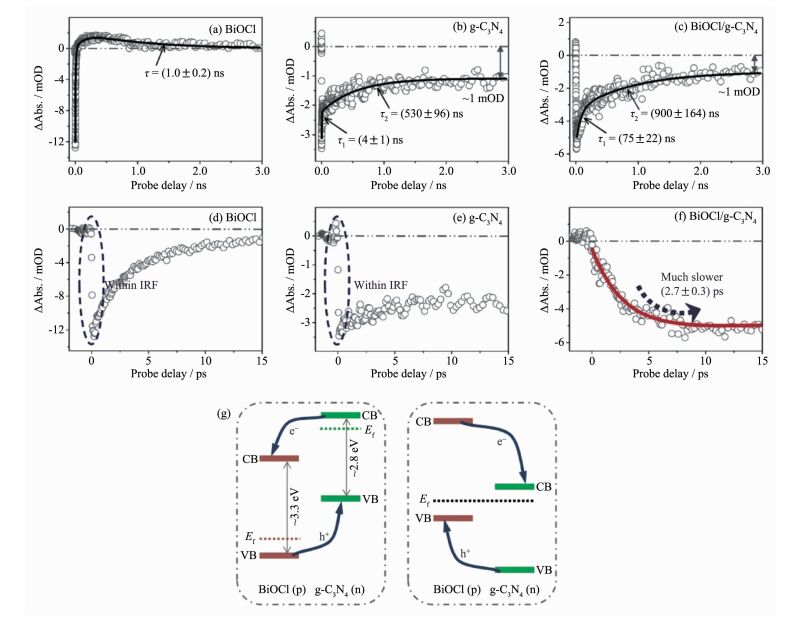
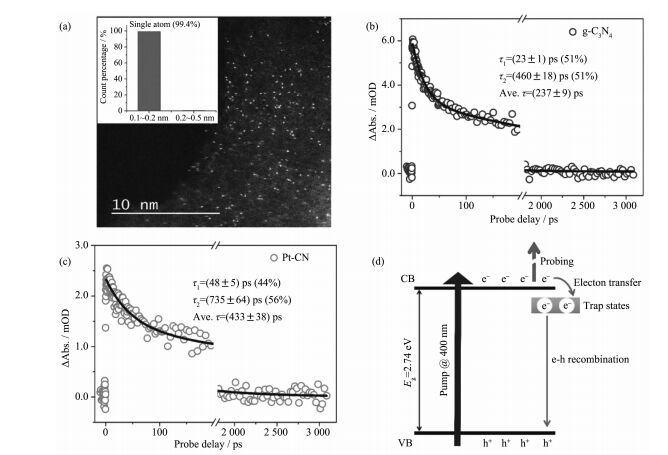
近年来,随着具有超快时间分辨、超高峰值功率的飞秒激光器的开发与应用,飞秒/皮秒时间分辨超快光谱技术为研究材料的中光生物种在不同时域范围内的动力学行为(例如光生物种的复合和分离、能量转移、激发态相干耦合等光物理过程)提供了可能,逐渐成为研究半导体材料光激发过程的最有效、直接的方法[27-31]。通常,利用超快光谱技术对光生物种的动力学过程进行研究,能够为材料光物理过程和光生物种弛豫机制的研究提供实验和理论指导。例如,Chen等利用超快光谱技术研究了氯氧化铋/聚合物氮化碳p-n结的界面电荷迁移问题(图 4),观测到了皮秒尺度的光生电子转移现象,明确了电子迁移方向为氯氧化铋至聚合物氮化碳[32];Wang等通过能流密度依赖的超快光谱测试,证实了聚合物氮化碳体系中强烈的三线态-三线态湮没现象,并在此基础上给出了完整的光物理模型,为其光激发过程研究提供了新思路[33]。
1.2 聚合物氮化碳材料的载流子和激子过程
目前,聚合物氮化碳材料光激发过程研究主要集中在光生载流子行为研究方面。通常,能带理论被用来理解光激发下聚合物氮化碳材料的载流子过程:如图 5a所示,在能量大于半导体能隙的光子激发下,体系中处于基态的电子会跃迁到导带上,并在价带上留下带正电的空穴;在经过快速的带内弛豫过程,光生电子和空穴分别在导带和价带内重新分布;光生载流子能够通过电子-空穴复合方式,使处于激发态的体系重新弛豫至基态。其中,光生载流子以辐射形式弛豫时,即发生光致发光现象。此外,这种处于激发态的电子和空穴(光生载流子)具有一定的还原和氧化能力,在迁移至材料表面后,能与外界离子或分子结合,实现光致电荷转移(即光催化、光电化学原理)。
然而,能带理论在研究聚合物氮化碳材料光激发过程中仍存在一定的局限性。这是由于能带理论是一种单电子近似理论,将单一电子所受其它电子的库仑作用,以及考虑电子波函数反对称性而带来的交换作用看作一个平均的等效的势场(晶格周期性势场)。虽然这种近似大大简化了求解过程,但是却忽略了某些情况下的体系中的多体作用。具体来说,能带理论忽略了半导体光激发产生的导带电子和价带空穴之间可能存在的相互作用,而将二者视为自由载荷子进而分别考虑它们的行为。这种近似对于大多数半导体材料来说影响较小。然而,在特定体系中(例如具有低介电常数的聚合物半导体材料),这种被长期忽略的光生载流子之间的相互作用将会影响甚至主导材料的光激发过程。一旦考虑到这种电子-空穴相互作用,就需要牵涉到一种不同于自由载流子的光生物种激子。
激子效应对半导体中的光吸收、发光和非线性光学性质起到至关重要的作用,在其光激发过程相关应用中具有重要意义[34-39]。由于具有较小的介电常数和较弱的屏蔽效应,聚合物半导体材料中光生载流子之间存在着潜在的强相互作用,而一旦考虑到这种潜在的相互作用,激子可能将会取代自由载流子成为主要的光生物种,对材料光激发过程及其应用产生重要影响。
首先,不同于基于载流子的电荷转移过程,激子能够通过共振传能方式实现能量的传递(如图 5b)[33]。以Förster型传能过程为例,该过程主要依赖偶极-偶极作用来实现催化剂与反应物之间的远距离能量传输,而不依赖于载流子(电子或空穴)的转移,通常来说,当供体与受体的激子能量及自旋匹配时,激子传能过程即可发生,且效率普遍高于基于载流子的电荷转移过程[40-41]。其次,考虑到与载流子过程之间的竞争机制,激子过程将成为限制载流子过程的重要潜在因素,同时材料的多体效应会导致光生物种间的强相互作用,会对光激发过程产生重要影响。不仅如此,材料的激子过程一直被认为会与体系光激发量子效率有关,为设计高效光功能材料提供了研究新思路[42-44]。因此,在研究聚合物氮化碳材料光激发过程中必须要考虑激子效应产生的影响。
近期,Wang等结合光致发光谱和超快光谱技术,系统地研究了聚合物氮化碳材料的光激发过程[26]。研究人员通过对聚合物氮化碳进行光致发光光谱研究,首次发现了材料中由三线态-三线态湮没诱导产生的P型延迟荧光现象(图 3),这种由2个三线态激子碰撞生成一个单线态激子的过程说明材料的多体效应对其光激发过程有显著影响;通过超快光谱分析对材料的激子动力学过程进行了研究,并结合发光数据首次完整的刻画了聚合物氮化碳的激子相关光物理模型(图 6),并通过改变能流密度和引入重原子等实验对模型的合理性进行了验证;并结合光催化实验结果,明确的指出了三线态-三线态湮没过程是限制聚合物氮化碳材料光激发物种(包括载流子和激子)浓度的决定因素。该项工作不仅为聚合物氮化碳材料的光激发过程研究提供指导,同时还为高效聚合物光功能材料的设计指明了新方向。
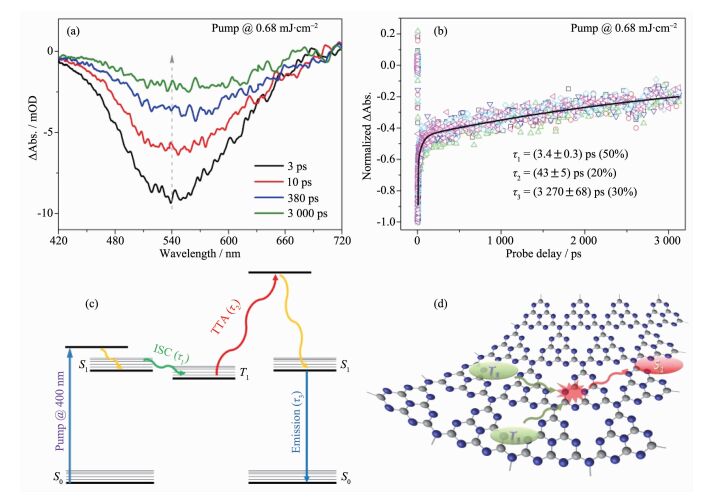 图6
聚合物氮化碳材料的激子过程研究: (a)延迟时间依赖瞬态吸收谱的演化; (b)瞬态吸收动力学衰减;
(c)相应的光物理模型; (d)材料中三线态-三线态湮没过程示意图[26]
Figure6.
(a) Representative transient absorption spectra at different probe delays and (b) corresponding kinetic traces at different probing wavelengths for g-C3N4; Schematic illustrations of (c) the photophysical processes and (d) triplet-triplet annihilation in the g-C3N4 system[26]
图6
聚合物氮化碳材料的激子过程研究: (a)延迟时间依赖瞬态吸收谱的演化; (b)瞬态吸收动力学衰减;
(c)相应的光物理模型; (d)材料中三线态-三线态湮没过程示意图[26]
Figure6.
(a) Representative transient absorption spectra at different probe delays and (b) corresponding kinetic traces at different probing wavelengths for g-C3N4; Schematic illustrations of (c) the photophysical processes and (d) triplet-triplet annihilation in the g-C3N4 system[26]
2 聚合物氮化碳的光激发过程调控
结合上述讨论,我们能够看出聚合物氮化碳光激发过程受材料的光吸收,能带结构,光生电荷分离效率,电荷迁移率,激子效应等特性的影响。在理解上述影响因素的作用机制基础上,可采取相应策略实现对聚合物氮化碳光激发过程的调控。这里,我们分别从载流子过程和激子过程的角度,对聚合物氮化碳的光激发过程调控研究进展进行总结。
2.1 载流子过程调控
由上面的描述可以看出一些能够影响体系光激发过程的因素。首先,材料的能带结构对于光激发过程有着至关重要的影响:一方面,能带结构中的禁带宽度决定着半导体的光吸收性质,一般来说,只有高于带隙能量的光子才能实现半导体的有效激发,进而生成自由电子和空穴,当禁带宽度过大时,会减小材料光吸收区间,不利于材料对太阳能的利用;另一方面,能带结构中价带和导带的能级位置决定光生空穴和电子的氧化还原能力,也就是说,只有价带和导带的能级位置合适时,才能使得光生载流子与外界物质产生光致电荷转移。此外,半导体中光生载流子行为特性也会对半导体光催化剂的性能产生重要影响。鉴于只有迁移至材料表面的光生载流子才有可能与外界物质产生光致电荷转移,光生电子和空穴的分离效率和迁移率将会直接影响材料的光响应性能;而电子和空穴可能会以非辐射形式复合,对材料发光性能和光致电荷转移效率产生影响。在上述理论和认识的基础上,可以得出能带结构调整和载流子行为优化能够有效调控材料的光激发过程。
目前,聚合物氮化碳材料的光激发过程调控研究主要集中在能带结构调整和载流子行为优化方面,建立起包括元素掺杂、结构无序度调节、维度控制、异质结的构建等多种调控策略[2-8]。
元素掺杂策略能够有效调节材料的电子结构,被广泛地应用于聚合物氮化碳的光激发过程调控。通常,铁、钛、锌等金属离子可以通过与氮原子键连方式,掺杂进聚合物氮化碳结构中[45-49]。得益于聚合物氮化碳均三嗪结构,体系的金属掺杂过程中主要通过金属离子与氮空中的氮原子形成强配位键,能够在保持聚合物氮化碳结构的同时,实现体系单原子级分散的金属杂原子的引入。一般认为,这种金属掺杂能够向能带结构中引入缺陷态,并通过捕获载流子而抑制体系中光生电子和空穴的复合。例如,Li等设计了一种单原子铂掺杂的聚合物氮化碳体系,并通过超快光谱技术证实了铂原子的引入能够显著改变材料表面陷阱态,光生电子的恢复时间显著增长(图 7)[50]。与金属元素的掺杂方式不同,氧、氮、磷、溴等元素则通过取代结构中碳、氮、氢原子的方式实现非金属掺杂,从而实现聚合物氮化碳的光激发过程的调控[51-55]。例如,Ma和Zhang等分别通过理论模拟和实验研究指出非金属掺杂能够有效提升结构载流子迁移率和光生载流子的分离效率[21, 56]。另外,研究人员还发现元素掺杂在促进载流子分离的同时,能够调控聚合物氮化碳材料的能带结构,实现体系光吸收的增强。中国科学技术大学熊宇杰教授课题组借鉴了均相配位化合物中金属与配体电荷转移过程诱导亚能带跃迁可以在低于能带能量范围内实现光吸收,通过利用聚合物氮化碳中氮原子作为配合位点,构建聚合物氮化碳/铂离子纳米配位结构,在铂含量低于千分之一的条件下即可实现体系拓展至红外波段的广谱吸光特性[57]。
针对其材料结构特点,研究人员提出了通过结构无序度控制来进行聚合物氮化碳能带结构和载流子行为的调控。例如,Ye等通过对聚合物氮化碳材料进行高于合成温度的热蚀刻处理,获得了具有高度结构扭曲的氮化碳样品,并通过理论模拟和实验研究证实了结构扭曲能够显著提升体系的n→π*跃迁,从而实现材料光吸收范围的拓展(图 8a,b)[58];Wang等提出利用体系结构无序度的改变,来调控聚合物氮化碳中的载流子行为,理论模拟显示,随着C-N面的扭曲,体系导带和价带的电荷密度呈现出明显的空间分离(图 8c),这一结果说明聚合物氮化碳材料的结构扭曲能够显著提高载流子分离效率,利于提升热载流子产率,基于此他们通过氟原子掺杂和热处理方法合成了一系列具有不同无序度的氮化碳材料,并利用电化学方法证实了体系无序度依赖的光生载流子分离现象(图 8d)[59]。值得说明的是,相较于元素掺杂方法,利用结构扭曲策略来调控材料光激发过程,能够避免引入杂质原子,利于实现聚合物氮化碳材料本征性质的研究。
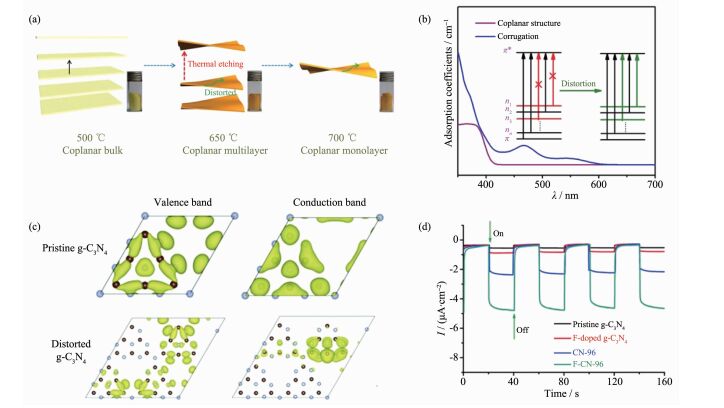 图8
结构无序度调控聚合物氮化碳能带结构和载流子行为: (a)高温热处理制备结构扭曲氮化碳样品和(b)理论计算模拟结构扭曲对于氮化碳材料光吸收特性的影响[58]; (c)理论模拟无序结构对于聚合物氮化碳能带结构的影响和(d)具有不同无序度聚合物氮化碳材料的光电流瞬态响应曲线[59]
Figure8.
(a) Schematic illustration of designing distorted polymeric carbon nitride nanosheets and (b) calculated theoretical optical absorption of coplanar and corrugation structures[58]; (c) Kohn-Sham orbitals for the valence and conduction bands of pristine g-C3N4 and structural distorted g-C3N4, respectively; (d) Photocurrent responses of samples with different degrees of distortion[59]
图8
结构无序度调控聚合物氮化碳能带结构和载流子行为: (a)高温热处理制备结构扭曲氮化碳样品和(b)理论计算模拟结构扭曲对于氮化碳材料光吸收特性的影响[58]; (c)理论模拟无序结构对于聚合物氮化碳能带结构的影响和(d)具有不同无序度聚合物氮化碳材料的光电流瞬态响应曲线[59]
Figure8.
(a) Schematic illustration of designing distorted polymeric carbon nitride nanosheets and (b) calculated theoretical optical absorption of coplanar and corrugation structures[58]; (c) Kohn-Sham orbitals for the valence and conduction bands of pristine g-C3N4 and structural distorted g-C3N4, respectively; (d) Photocurrent responses of samples with different degrees of distortion[59]
进一步,考虑到聚合物氮化碳材料的层状结构特点,研究人员还提出通过维度控制来调控材料的载流子行为[20, 60-63]。例如,Zhang等通过密度泛函理论计算研究了聚合物氮化碳维度对其电子结构的影响,指出随着材料维度的降低,C-N面内的碳与氮原子之间的杂化逐渐增强,使得导带边态密度的显著增强,预示着二维氮化碳材料是一种潜在的发光材料,并通过水相剥离方法,制备了具有高效光响应性能的超薄聚合物氮化碳纳米片,其发光量子效率高达19.6%,较之相应块材提升了大约4倍,如图 9(a,b)所示[20];此外,他们还进一步证实了通过改良液相剥离法获得了具有优异的双光子吸收特性的氮化碳量子点,在低能量的红光激发下,产生高能量的绿光发射,在750 nm光激发下,双光子吸收截面高达28 000 GM(GM为双光子吸收截面单位,1 GM=1×10-50 cm4·s·molecule-1·photon-1),并通过理论模拟得出其双光子吸收特性是与单层氮化碳结构中增强的C-N面内共轭载流子π-π*电荷转移过程有关,如图 9(c~e)所示[60]。
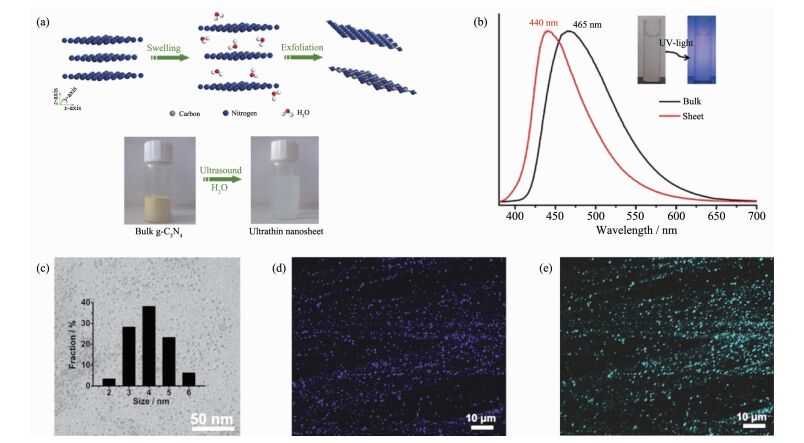 图9
维度控制来调控聚合物氮化碳材料的载流子行为: (a)液相剥离法制备超薄氮化碳纳米片示意图和(b)聚合物氮化碳纳米片和相应块材的荧光发射谱图[20]; (c)氮化碳量子点的透射图像及尺寸分布及其(d)单光子和(e)双光子荧光成像[60]
Figure9.
(a) Schematic illustration of liquid-exfoliation process from bulk g-C3N4 to corresponding ultrathin nanosheets;
(b) Normalized photoluminescence spectra of bulk and ultrathin g-C3N4 nanosheets. Inset: the photographs of ultrathin g-C3N4 nanosheets solution without/with UV light illumination[20]; (c) TEM image and corresponding size distribution of the obtained g-C3N4 quantum dots and (d) One-photon and (e) two-photon luminescence image of g-C3N4 quantum dots dispersed in a glass substrate, respectively[60]
图9
维度控制来调控聚合物氮化碳材料的载流子行为: (a)液相剥离法制备超薄氮化碳纳米片示意图和(b)聚合物氮化碳纳米片和相应块材的荧光发射谱图[20]; (c)氮化碳量子点的透射图像及尺寸分布及其(d)单光子和(e)双光子荧光成像[60]
Figure9.
(a) Schematic illustration of liquid-exfoliation process from bulk g-C3N4 to corresponding ultrathin nanosheets;
(b) Normalized photoluminescence spectra of bulk and ultrathin g-C3N4 nanosheets. Inset: the photographs of ultrathin g-C3N4 nanosheets solution without/with UV light illumination[20]; (c) TEM image and corresponding size distribution of the obtained g-C3N4 quantum dots and (d) One-photon and (e) two-photon luminescence image of g-C3N4 quantum dots dispersed in a glass substrate, respectively[60]
另外,研究人员还通过构建异质结结构来调控聚合物氮化碳材料的光激发过程[63-67]。通常,由于不同材料之间能带结构的差异,能够使得异质结界面处的光生载流子向低能级迁移,当材料能带结构合适时,可以实现电子和空穴在复合材料中不同半导体上富集,从而抑制光生载流子复合。除此之外,异质结结构还可能会对载流子迁移率,能量传递机制等产生影响。
上述对聚合物氮化碳材料的能带结构和载流子行为调控的策略,为基于载流子过程的光激发应用及其性能优化提供了材料基础。
2.2 激子过程调控
基于对其材料结构和激子过程的理解,Wang等首次提出通过羰基官能团的引入来调控聚合物氮化碳材料中三线态激子产率[33]。如图 10所示,研究人员通过氧化性酸处理向聚合物氮化碳材料的中引入羰基官能团,并通过发光光谱实验证实羰基能够协同地增强体系自旋-轨道耦合和降低单线态-三线态能隙,从而促进体系的系间窜越效率,最终实现较之原材料显著提升的三线态激子产率。
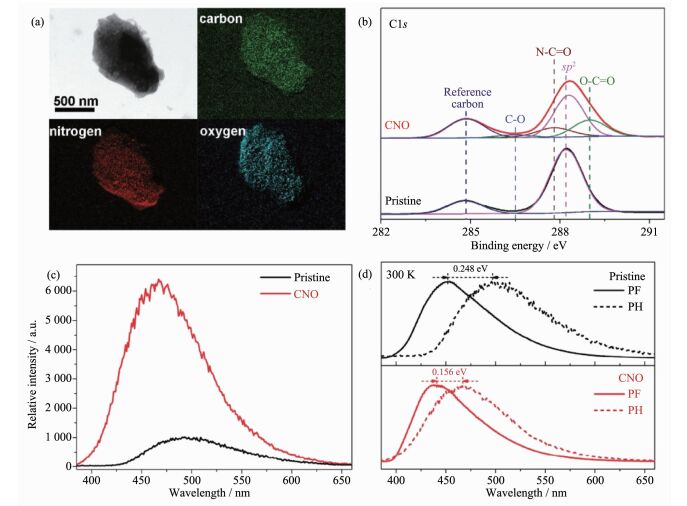 图10
(a) 氮化碳氧化物的HAADF-STEM图和相应元素成像; 氮化碳氧化物和聚合物氮化碳原材料的(b) C1s X射线光电子能谱; (c)磷光光谱和(d)强度归一化的荧光/磷光光谱[33]
Figure10.
(a) Elemental mapping images of CNO; (b) XPS C1s spectra, (c) phosphorescence (PH) spectra and (d) normalized steady-state prompt fluorescence (PF) and PH spectra of pristine g-C3N4 and CNO, respectively[33]
图10
(a) 氮化碳氧化物的HAADF-STEM图和相应元素成像; 氮化碳氧化物和聚合物氮化碳原材料的(b) C1s X射线光电子能谱; (c)磷光光谱和(d)强度归一化的荧光/磷光光谱[33]
Figure10.
(a) Elemental mapping images of CNO; (b) XPS C1s spectra, (c) phosphorescence (PH) spectra and (d) normalized steady-state prompt fluorescence (PF) and PH spectra of pristine g-C3N4 and CNO, respectively[33]
值得说明的是,光激发过程中的激子行为和载流子行为并不是独立、不相关的,而是相互依赖、相互竞争的关系。也就是说任何调控方法,都不可避免的对2种光生物种的行为产生影响。例如,如图 11(a,b)所示,Wang等通过引入溴原子增强聚合物氮化碳体系旋轨耦合,来促进三线态浓度的增强,从而提升三线态-三线态湮没过程;然而,如图 11(c,d)所示,实验结果显示这种条件下不论载流子还是激子都存在较大消耗,不仅证实了加速的三线态-三线态湮没严重限制了材料光激发物种(包括载流子和激子)的浓度,同时还说明体系中载流子和激子之间存在着快速的相互转化; 此外,聚合物氮化碳纳米片的发光光谱证实其高的单线态产率与被抑制的三线态-三线态湮没过程有关[26]。这种相关联的激子行为和载流子行为提示我们,在研究材料光激发过程调控时,需要全面考虑调控策略对二者产生的影响。
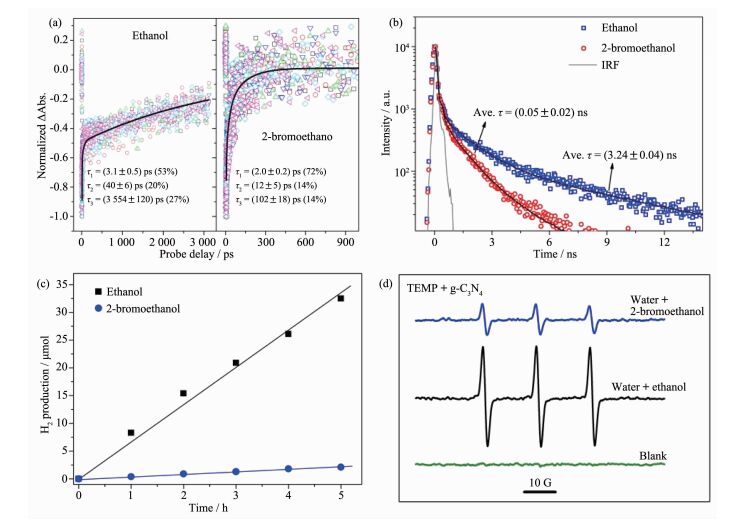 图11
溴原子对聚合物氮化碳体系光激发过程的影响: (a)瞬态吸收动力学, (b)时间分辨荧光光谱,
(c)光催化产氢性能和(d)单线态氧ESR捕获实验[26]
Figure11.
(a) Representative transient absorption kinetic traces, (b) time-resolved prompt fluorescence kinetics,
(c) time-dependent photocatalytic H2 evolution, and (d) TEMP-trapped ESR measurements with/without the addition of bromine atoms[26]
图11
溴原子对聚合物氮化碳体系光激发过程的影响: (a)瞬态吸收动力学, (b)时间分辨荧光光谱,
(c)光催化产氢性能和(d)单线态氧ESR捕获实验[26]
Figure11.
(a) Representative transient absorption kinetic traces, (b) time-resolved prompt fluorescence kinetics,
(c) time-dependent photocatalytic H2 evolution, and (d) TEMP-trapped ESR measurements with/without the addition of bromine atoms[26]
此外,鉴于光激发过程中激子与热载荷子的竞争关系,可以通过促进激子解离来实现体系载流子浓度的调控。基于这一观点,Wang等通过离子辅助溶剂热法合成出半结晶氮化碳,利用稳态和时间分辨荧光/磷光光谱其结构中丰富的有序-无序界面来促进体系中单线态/三线态激子的解离(图 12);结合理论计算,他们指出有序链较之无序链具有更低的最高占有轨道能级和最低空轨道能级,这一能级特点预示着解离产生的电子和空穴分别向有序链和无序链注入,而2种链共轭程度的差异,导致了进一步的电子迁出和空穴阻断,电化学测试结果显示半结晶氮化碳样品中的电子浓度和光电流分别是原材料的7倍和5倍[68]。
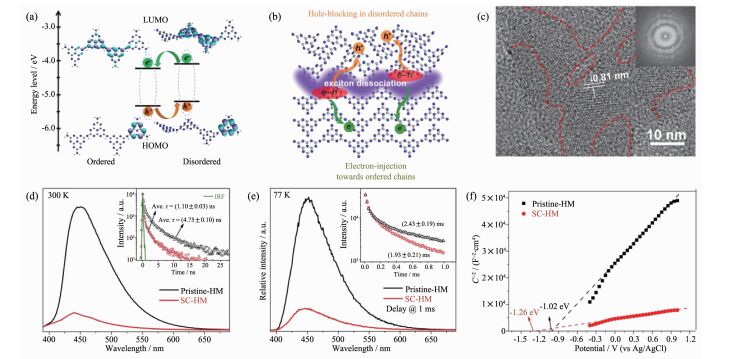 图12
(a) 有序和无序三均三嗪链的密度泛函理论计算; (b)半结晶三均三嗪链中激子解离和电荷转移示意图;
(c)离子辅助溶剂热法合成半结晶氮化碳样品的高分辨透射电镜图像; 半结晶氮化碳及其原材料的(d) 300 K下稳态荧光光谱, (e)稳态延迟荧光光谱, (f) Mott-Schottky曲线[68]
Figure12.
(a) DFT simulations of ordered and disordered heptazine-based chains; (b) Schematic illustration of exciton dissociation and charge transfer in semicrystalline heptazine-based melon; (c) High-resolution electron-microscope image of semicrystalline heptazine-based melon, inset: corresponding Fourier transform pattern; Steady-state and time-resolved (d) fluorescence and delayed fluorescence spectra, (e) Mott-Schottky curves of pristine-HM and SC-HM samples, respectively[68]
图12
(a) 有序和无序三均三嗪链的密度泛函理论计算; (b)半结晶三均三嗪链中激子解离和电荷转移示意图;
(c)离子辅助溶剂热法合成半结晶氮化碳样品的高分辨透射电镜图像; 半结晶氮化碳及其原材料的(d) 300 K下稳态荧光光谱, (e)稳态延迟荧光光谱, (f) Mott-Schottky曲线[68]
Figure12.
(a) DFT simulations of ordered and disordered heptazine-based chains; (b) Schematic illustration of exciton dissociation and charge transfer in semicrystalline heptazine-based melon; (c) High-resolution electron-microscope image of semicrystalline heptazine-based melon, inset: corresponding Fourier transform pattern; Steady-state and time-resolved (d) fluorescence and delayed fluorescence spectra, (e) Mott-Schottky curves of pristine-HM and SC-HM samples, respectively[68]
3 聚合物氮化碳光激发过程的功能化
3.1 光催化特性研究
自2009年王心晨教授等首次报道了其具有光催化产氢性质[1]以来,聚合物氮化碳材料在光催化领域的应用激发了研究人员极大的兴趣。经过诸多努力,研究人员不仅将其催化研究拓展至污染物降解、二氧化碳还原、有机物选择性氧化、固氮等众多反应中,还在材料催化反应机制研究和光催化剂设计方面取得了显著成果[2-6]。例如,王心晨教授课题组和成会明院士课题组分别通过液相剥离和氧蚀刻方法制备了聚合物氮化碳纳米片,呈现出较之原材料显著提升的产氢性能[61-62];上海交通大学李新昊研究员系统研究了异质结结构对于聚合物氮化碳光催化性能的影响,设计出一系列具有优异有机催化性能的聚合物氮化碳基催化剂[69-71];Lu等利用亚纳米孔修饰和钛氧原子掺杂策略对聚合物氮化碳纳米片电子结构进行调控,使得体系光生载流子分离效率和电荷迁移速率得到促进,实现了材料光催化性能的提升[72]。
需要指出的是,当前聚合物氮化碳光催化水分解反应的研究大多集中在产氢或者产氧半反应的研究方面[73],并且在催化过程中牵涉到牺牲剂的使用。这不仅限制了催化剂的实际应用,还阻碍了材料本征光催化机制的研究,因而,实现光催化水分子全解已成为研究人员追求的目标。近期,苏州大学康振辉教授课题组提出了一种两步、两电子过程的催化机制,他们通过设计了一种碳纳米点/氮化碳复合光催化体系,利用中间产物过氧化氢的转化,实现了可见光下高效的全解水反应,在λ=(420±20) nm光照条件下,量子效率可达16%,而体系的太阳能转化效率约为2%(图 13)[74]。
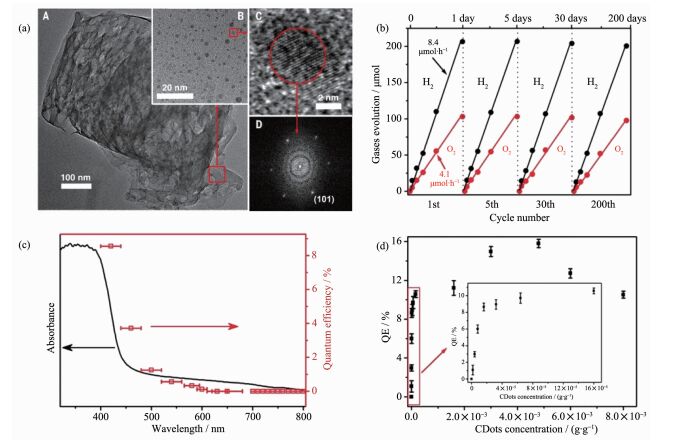 图13
聚合物氮化碳/碳量子点材料的(a)形貌表征; (b)可见光下的光催化产氢和产氧量; (c)波长依赖的水分解量子效率; (d)碳量子点浓度依赖的水分解量子效率[74]
Figure13.
(a) Characterization of the physical structure of CDots-C3N4; (b) Time dependent H2 and O2 production under visible light irradiation of CDots-C3N4; (c) Wavelength dependent quantum efficiency of water splitting of CDots-C3N4;
(d) Quantum efficiency for different concentrations of CDots in composite catalyst[74]
图13
聚合物氮化碳/碳量子点材料的(a)形貌表征; (b)可见光下的光催化产氢和产氧量; (c)波长依赖的水分解量子效率; (d)碳量子点浓度依赖的水分解量子效率[74]
Figure13.
(a) Characterization of the physical structure of CDots-C3N4; (b) Time dependent H2 and O2 production under visible light irradiation of CDots-C3N4; (c) Wavelength dependent quantum efficiency of water splitting of CDots-C3N4;
(d) Quantum efficiency for different concentrations of CDots in composite catalyst[74]
目前,聚合物氮化碳光催化研究主要关注基于热载流子电荷传递过程的反应。在这一过程中,催化剂和反应物之前存在明显的电荷净转移,二者反应前后带电量出现改变,同时该过程要求二者能级结构合适,即存在反应物的氧化/还原过程。例如,光催化分解水反应中,水分子与催化剂之间存在电荷转移过程,水分子被光生电子还原成氢气,同时被空穴氧化成氧气。除了上述这些基于热载流子电荷传递过程的反应,Wang等近期首次研究了聚合物氮化碳材料中基于激子传能过程的光催化应用[33]。研究人员以氧分子活化反应为例,考察了激子过程对聚合物氮化碳光催化过程的影响,通过利用具有提升的三线态产率的羰基化氮化碳,促进氧分子活化反应中的基于激子传能过程的单线态氧产生,同时极大地抑制了电荷传递过程相关的其他活性氧物种的生成,这种高选择性的活性氧物种产生赋予材料良好的选择性氧化能力,如图 14(a,b)所示;此外,他们还进一步利用有序-无序界面来进行激子过程调控,增强体系载流子相关光催化性能,结果显示,半结晶氮化碳材料具有显著提升的激子解离和热载流子的产生,在提升热电子相关的超氧自由基生成的同时,抑制了单线态氧产生,如图 14(c,d)所示[68]。
 图14
氮化碳氧化物及相应块材的(a)气氛依赖的四甲基联苯胺氧化实验和(b)单线态氧ESR捕获测试[33]; 半结晶氮化碳及相应原材料的(c)超氧自由基ESR捕获测试和(d)苯甲醇氧化实验[68]
Figure14.
(a) Evaluation of TMB oxidation under different gas conditions and (b) ESR spectra for singlet oxygen detection with pristine g-C3N4 and CNO[33]; (c) ESR spectra of pristine-HM and SC-HM samples in the presence of DMPO in methanol and (d) wavelength-dependent benzyl alcohol oxidation with pristine-HM and SC-HM sample, respectively[68]
图14
氮化碳氧化物及相应块材的(a)气氛依赖的四甲基联苯胺氧化实验和(b)单线态氧ESR捕获测试[33]; 半结晶氮化碳及相应原材料的(c)超氧自由基ESR捕获测试和(d)苯甲醇氧化实验[68]
Figure14.
(a) Evaluation of TMB oxidation under different gas conditions and (b) ESR spectra for singlet oxygen detection with pristine g-C3N4 and CNO[33]; (c) ESR spectra of pristine-HM and SC-HM samples in the presence of DMPO in methanol and (d) wavelength-dependent benzyl alcohol oxidation with pristine-HM and SC-HM sample, respectively[68]
3.2 光致发光特性研究
鉴于其高效的荧光发射特性和良好的生物兼容性,聚合物氮化碳材料在生物成像等方面具有重要的应用价值[20, 60]。Zhang等通过水相剥离的方法制备了具有良好水溶性、尺寸分布在100 nm左右、高效发光特性的超薄聚合物氮化碳纳米片,并将其应用于细胞成像,实验结果表明,所获得的氮化碳纳米片能够有效的吸附在细胞膜的表面,实现对细胞膜的特异性成像,性能可比拟商业品荧光染料(图 15A)[20];进一步研究发现,改良液相剥离法获得的单层量子点能够呈现出远优于传统的有机染料分子的双光子吸收特性,而得益于材料合适的尺寸分布以及良好的DNA结合特性,聚合物氮化碳量子点能够特异性的对细胞核进行双光子成像(图 15B)[60]。
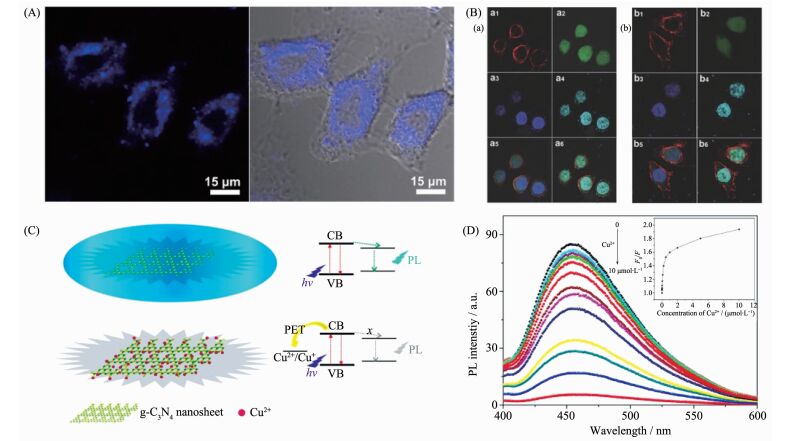 图15
(A) 聚合物氮化碳纳米片的细胞膜成像实验[20]; (B)聚合物氮化碳量子点对细胞核特异性成像[60]; (C)聚合物氮化碳纳米片的铜离子检测原理图和(D)不同铜离子浓度下,聚合物氮化碳纳米片的荧光谱图[75]
Figure15.
(A) Confocal fluorescence image and overlay image of bright field and confocal fluorescence image of the HeLa cells incubated with ultrathin g-C3N4 nanosheets[20]; (B) One-photon and two-photon confocal fluorescent images of HepG2 cells co-stained with (a) DiI, FDA, g-C3N4 QDs, and (b) DiI, FDA, DAPI, respectively[60]; (C) Schematic illustration of the g-C3N4 nanosheets-based fluorosensor for Cu2+ and (D) PL spectra of g-C3N4 nanosheets dispersion in the presence of different Cu2+ concentrations[75]
图15
(A) 聚合物氮化碳纳米片的细胞膜成像实验[20]; (B)聚合物氮化碳量子点对细胞核特异性成像[60]; (C)聚合物氮化碳纳米片的铜离子检测原理图和(D)不同铜离子浓度下,聚合物氮化碳纳米片的荧光谱图[75]
Figure15.
(A) Confocal fluorescence image and overlay image of bright field and confocal fluorescence image of the HeLa cells incubated with ultrathin g-C3N4 nanosheets[20]; (B) One-photon and two-photon confocal fluorescent images of HepG2 cells co-stained with (a) DiI, FDA, g-C3N4 QDs, and (b) DiI, FDA, DAPI, respectively[60]; (C) Schematic illustration of the g-C3N4 nanosheets-based fluorosensor for Cu2+ and (D) PL spectra of g-C3N4 nanosheets dispersion in the presence of different Cu2+ concentrations[75]
除了细胞成像以外,聚合物氮化碳的发光特性还被应用于金属离子、小分子等的检测[75-78]。如图 15(C,D)所示,Tian等利用铜离子与聚合物氮化碳纳米片之间光生电子转移导致的纳米片的荧光淬灭现象,成功的将材料作为荧光传感器应用于选择性铜离子检测,检测限浓度低至0.1 nmol·L-1,并且在其他金属离子存在条件下,检测结果不会产生明显变化[75];而Zhang等则利用谷胱甘肽抑制了聚合物氮化碳/二氧化锰体系的荧光淬灭现象,使得体系荧光发射的增强,实现了最低浓度0.2 μmol·L-1的谷胱甘肽检测[76];Wang等利用具有荧光基团标记的DNA作为探针,研究了聚合物氮化碳纳米片与DNA的相互作用能力,利用光致电荷转移导致的荧光淬灭现象,发展了用于快速DNA荧光检测方法[77]。
3.3 光电化学特性研究
考虑其良好的光响应性质以及优异的材料物性,聚合物氮化碳的光电化学性质[79-82]的研究同样吸引了科研人员的注意,在光电催化和光电化学传感等方面取得了系列成果。
通常,由于聚合物氮化碳材料较差的电荷迁移率,研究人员通常利用构建复合结构来提升体系的光电催化性能。Su等利用磷掺杂的聚合物氮化碳修饰二氧化钛纳米管,以增强催化剂的光吸收、提高材料电导率和促进光生载流子的分离,实现0 V偏压下(vs Ag/AgCl,pH=6.6)较之未修饰电极大约提升3倍的光电流[83];如图 16(a,b)所示,Hou等设计了一种具有特殊分枝结构的三氧化钨/聚合物氮化碳/钴氧化物的纳米催化剂,得益于复合材料的三维小尺寸结构导致的增强光吸收、异质结诱导的电荷分离以及高比表面造成的快速界面电荷收集和表面反应,该体系呈现出优异的光电催化水氧化性能[84]。
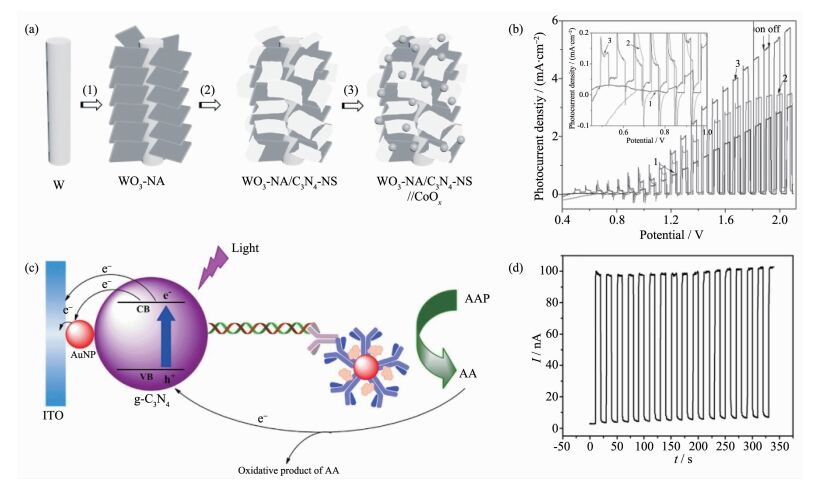 图16
具有分枝结构三氧化钨/聚合物氮化碳/钴氧化物纳米催化剂的(a)合成示意图及其(b)光电催化水氧化性能[84]; 基于聚合物氮化碳/金纳米颗粒光电极的小分子核糖核酸检测(c)机理图及其(d)光电流响应测试[85]
Figure16.
(a) Synthetic route to the three-dimensionally branched WO3 nanosheet array /C3N4 nanosheets//CoOx, and (b) Variation of photocurrent density versus applied voltage of different samples[84]; (c) MicroRNA-319a detection mechanism with g-C3N4-AuNPs nanohybrids and (d) Time-based photocurrent responses of the biosensor[85]
图16
具有分枝结构三氧化钨/聚合物氮化碳/钴氧化物纳米催化剂的(a)合成示意图及其(b)光电催化水氧化性能[84]; 基于聚合物氮化碳/金纳米颗粒光电极的小分子核糖核酸检测(c)机理图及其(d)光电流响应测试[85]
Figure16.
(a) Synthetic route to the three-dimensionally branched WO3 nanosheet array /C3N4 nanosheets//CoOx, and (b) Variation of photocurrent density versus applied voltage of different samples[84]; (c) MicroRNA-319a detection mechanism with g-C3N4-AuNPs nanohybrids and (d) Time-based photocurrent responses of the biosensor[85]
聚合物氮化碳还可用于光电化学免疫测定生物分子,基本原理是光激发下,聚合物氮化碳光电极和目标分子之间的光致电荷转移引起的光电流信号的改变。研究人员一般通过异质结结构的建立来促进体系载流子分离效率,从而提升检测灵敏度。例如,如图 16(c,d)所示,Yin等通过选择聚合物氮化碳/金纳米颗粒作为光活性材料、Anti-DNA:RNA抗体作为目标分子识别单元、抗坏血酸2-磷酸盐作为电子给体,实现了小分子核糖核酸的灵敏选择性检测[85]。此外,还有关于基于聚合物氮化碳体系的蛋白激酶、多核苷酸激酶等光电化学检测的报道[86-89]。
4 结论与展望
作为一类新兴的半导体材料,聚合物氮化碳优异的光响应特性使得材料在光催化、光致发光、光电化学等领域具有重要的应用前景。在实现材料性能最优化的探索方面,聚合物氮化碳光激发过程研究吸引了越来越多的注意。近些年,随着表征技术的发展,相关光激发过程研究取得了丰富的成果,不仅加深了对材料光响应特性的理解,也为材料的优化设计提供了理论和实验指导。在前期工作基础上,我们从载流子和激子的观点上,综述了近期聚合物氮化碳材料光激发过程研究、调控及应用拓展方面取得的进展。然而,受限于材料结构和表征手段等因素,较之经典半导体体系,聚合物氮化碳材料的光激发过程研究依旧存在诸多问题,这为今后更深入的研究提供了参考与目标。未来,聚合物氮化碳体系研究突破点将集中在精准结构的设计,同时需要全面考虑光生物种之间的相互作用对其光激发过程的影响。
-
-
[1]
Wang X, Maeda K, Thomas A, et al. Nat. Mater., 2009, 8:76-80 doi: 10.1038/nmat2317
-
[2]
张金水, 王博, 王心晨.化学进展, 2014, 26 (1):19-29ZHANG Jin-Shui, WANG Bo, WANG Xin-Chen. Progress in Chemistry, 2014, 26 (1):19-29
-
[3]
Wang X, Blechert S, Antonietti M. ACS Catal., 2012, 2:1596-1606 doi: 10.1021/cs300240x
-
[4]
Ong W J, Tan L L, Ng Y H, et al. Chem. Rev., 2016, 116: 7159-7329 doi: 10.1021/acs.chemrev.6b00075
-
[5]
Zhang J, Chen Y, Wang X. Energy Environ. Sci., 2015, 8: 3092-3108 doi: 10.1039/C5EE01895A
-
[6]
Cao S, Yu J. J. Phys. Chem. Lett., 2014, 5:2101-2107 doi: 10.1021/jz500546b
-
[7]
Cao S, Low J, Yu J, et al. Adv. Mater., 2015, 27:2150-2176 doi: 10.1002/adma.201500033
-
[8]
Liu J, Wang H, Antonietti M. Chem. Soc. Rev., 2016, 45: 2308-2326 doi: 10.1039/C5CS00767D
-
[9]
Thomas A, Fischer A, Goettmann F, et al. J. Mater. Chem., 2008, 18:4893-4908 doi: 10.1039/b800274f
-
[10]
Kroke E, Schwarz M, Horath-Bordon E, et al. New J. Chem., 2002, 26:508-512 doi: 10.1039/b111062b
-
[11]
Sattler A, Pagano S, Zeuner M, et al. Chem.-Eur. J., 2009, 15:13161-13170 doi: 10.1002/chem.v15:47
-
[12]
Lotsch B V, Schnick W. Chem. Mater., 2006, 18:1891-1900 doi: 10.1021/cm052342f
-
[13]
Redemann C E, Lucas H J. J. Am. Chem. Soc., 1940, 62: 842-846 doi: 10.1021/ja01861a038
-
[14]
Ang T P, Chan Y M. J. Phys. Chem. C, 2011, 115:15965-15972 doi: 10.1021/jp200324v
-
[15]
Wei W, Jacob T. Phys. Rev. B, 2013, 87:085202 doi: 10.1103/PhysRevB.87.085202
-
[16]
Butchosa C, Guiglion P, Zwijnenburg M A. J. Phys. Chem. C, 2014, 118:24833-24842 doi: 10.1021/jp507372n
-
[17]
Zhang H, Zuo X, Tang H, et al. Phys. Chem. Chem. Phys., 2015, 17:6280-6288 doi: 10.1039/C4CP05288A
-
[18]
Dong G, Zhang Y, Pan Q, et al. J. Photochem. Photobiol. C, 2014, 20:33-50 doi: 10.1016/j.jphotochemrev.2014.04.002
-
[19]
Du A, Sanvito S, Smith S C. Phys. Rev. Lett., 2012, 108: 197207 doi: 10.1103/PhysRevLett.108.197207
-
[20]
Zhang X D, Xie X, Wang H, et al. J. Am. Chem. Soc., 2013, 135:18-21 doi: 10.1021/ja308249k
-
[21]
Ma X G, Lü Y H, Xu J, et al. J. Phys. Chem. C, 2012, 116: 23485-23493 doi: 10.1021/jp308334x
-
[22]
O'Connell M J, Bachilo S M, Huffman C B, et al. Science, 2002, 297:593-596 doi: 10.1126/science.1072631
-
[23]
Hill H M, Rigosi A F, Roquelet C, et al. Nano Lett., 2015, 15:2992-2997 doi: 10.1021/nl504868p
-
[24]
Liu K, Zhang L, Cao T, et al. Nat. Commun., 2014, 5:4966 doi: 10.1038/ncomms5966
-
[25]
Sun L, Chen Z, Ren Q, et al. Phys. Rev. Lett., 2008, 100: 156403 doi: 10.1103/PhysRevLett.100.156403
-
[26]
Wang H, Jiang S L, Chen S C, et al. Chem. Sci., 2017, 8: 4087-4092 doi: 10.1039/C7SC00307B
-
[27]
Zhang Q, Zheng H, Geng Z, et al. J. Am. Chem. Soc., 2013, 135:12468-12474 doi: 10.1021/ja407110r
-
[28]
Bi W, Li X, Zhang L, et al. Nat. Commun., 2015, 6:8647 doi: 10.1038/ncomms9647
-
[29]
Wu K, Chen J, McBride J R, et al. Science, 2015, 349:632-635 doi: 10.1126/science.aac5443
-
[30]
Sun X, Wang X, Li X, et al. Macromol. Rapid Commun., 2015, 36:298-303 doi: 10.1002/marc.201400529
-
[31]
Zhang Q, Luo Y. High Power Laser Sci. Eng., 2016, 4:e22 doi: 10.1017/hpl.2016.23
-
[32]
Chen Z W, Zhang Q, Luo Y. ChemPhotoChem, 2017, 1:350-354 doi: 10.1002/cptc.v1.8
-
[33]
Wang H, Jiang S, Chen S, et al. Adv. Mater., 2016, 28:6940-6945 doi: 10.1002/adma.201601413
-
[34]
Scholes G D, Rumbles G. Nat. Mater., 2006, 5:683-696 doi: 10.1038/nmat1710
-
[35]
Goushi K, Yoshida K, Sato K, et al. Nat. Photonics, 2012, 6: 253-258 doi: 10.1038/nphoton.2012.31
-
[36]
Huang M H, Mao S, Feick H, et al. Science, 2001, 292:1897-1899 doi: 10.1126/science.1060367
-
[37]
Xie Y, Gong M, Shastry T A, et al. Adv. Mater., 2013, 25: 3433-3437 doi: 10.1002/adma.v25.25
-
[38]
Nozik A J, Beard M C, Luther J M, et al. Chem. Rev., 2010, 110:6873-6890 doi: 10.1021/cr900289f
-
[39]
Semonin O E, Luther J M, Choi S, et al. Science, 2011, 334: 1530-1533 doi: 10.1126/science.1209845
-
[40]
Jares-Erijman E A, Jovin T M. Nat. Biotechnol., 2003, 21: 1387-1395 doi: 10.1038/nbt896
-
[41]
Sekar R B, Periasamy A. J. Cell Biol., 2003, 160:629-633 doi: 10.1083/jcb.200210140
-
[42]
Cao Y, Parker I D, Yu G, et al. Nature, 1999, 397:414-417 doi: 10.1038/17087
-
[43]
Congreve D N, Lee J, Thompson N J, et al. Science, 2013, 340:334-337 doi: 10.1126/science.1232994
-
[44]
Lee J, Jadhav P, Reusswig P D, et al. Acc. Chem. Res., 2013, 46:1300-1311 doi: 10.1021/ar300288e
-
[45]
Wang X, Chen X, Thomas A, et al. Adv. Mater., 2009, 21: 1609-1612 doi: 10.1002/adma.v21:16
-
[46]
Gao L F, Wen T, Xu J Y, et al. ACS Appl. Mater. Interfaces, 2016, 8:617-624 doi: 10.1021/acsami.5b09684
-
[47]
Yue B, Li Q, Iwai H, et al. Sci. Technol. Adv. Mater., 2011, 12:034401 doi: 10.1088/1468-6996/12/3/034401
-
[48]
Ding G, Wang W, Jiang T, et al. ChemCatChem, 2013, 5: 192-200 doi: 10.1002/cctc.201200502
-
[49]
Wang Y, Wang Y, Chen Y, et al. Mater. Lett., 2015, 139:70-72 doi: 10.1016/j.matlet.2014.10.008
-
[50]
Li X, Bi W, Zhang L, et al. Adv. Mater., 2016, 28:2427-2431 doi: 10.1002/adma.201505281
-
[51]
Fang J, Fan H, Li M, et al. J. Mater. Chem. A, 2015, 3:13819-13826 doi: 10.1039/C5TA02257F
-
[52]
Zhang G, Zhang M, Ye X, et al. Adv. Mater., 2014, 26:805-809 doi: 10.1002/adma.201303611
-
[53]
Wang Y, Li H, Yao J, et al. Chem. Sci., 2011, 2:446-450 doi: 10.1039/C0SC00475H
-
[54]
Wang Y, Di Y, Antonietti M, et al. Chem. Mater., 2010, 22: 5119-5121 doi: 10.1021/cm1019102
-
[55]
Liu G, Niu P, Sun C, et al. J. Am. Chem. Soc., 2010, 132: 11642-11648 doi: 10.1021/ja103798k
-
[56]
Zhang Y J, Mori T, Ye J H, et al. J. Am. Chem. Soc., 2010, 132:6294-6295 doi: 10.1021/ja101749y
-
[57]
Li Y, Wang Z, Xia T, et al. Adv. Mater., 2016, 28:6959-6965 doi: 10.1002/adma.201601960
-
[58]
Chen Y, Wang B, Lin S, et al. J. Phys. Chem. C, 2014, 118: 29981-29989 doi: 10.1021/jp510187c
-
[59]
Wang H, Zhang X D, Xie J, et al. Nanoscale, 2015, 7:5152-5156 doi: 10.1039/C4NR07645A
-
[60]
Zhang X, Wang H, Wang H, et al. Adv. Mater., 2014, 26: 4438-4443 doi: 10.1002/adma.v26.26
-
[61]
Yang S, Gong Y, Zhang J, et al. Adv. Mater., 2013, 25:2452-2456 doi: 10.1002/adma.v25.17
-
[62]
Niu P, Zhang L, Liu G, et al. Adv. Funct. Mater., 2012, 22: 4763-4770 doi: 10.1002/adfm.v22.22
-
[63]
Zhao Y, Zhang J, Qu L. ChemNanoMat, 2015, 1:298-318 doi: 10.1002/cnma.v1.5
-
[64]
Dong F, Zhao Z, Xiong T, et al. ACS Appl. Mater. Interfaces, 2013, 5:11392-11401 doi: 10.1021/am403653a
-
[65]
Samanta S, Martha S, Parida K. ChemCatChem, 2014, 6:1453-1462
-
[66]
Bi L, Xu D, Zhang L, et al. Phys. Chem. Chem. Phys., 2015, 17:29899-29905 doi: 10.1039/C5CP05158D
-
[67]
Zhao Z, Sun Y, Dong F. Nanoscale, 2015, 7:15-37 doi: 10.1039/C4NR03008G
-
[68]
Wang H, Sun X, Li D, et al. J. Am. Chem. Soc., 2017, 139: 2468-2473 doi: 10.1021/jacs.6b12878
-
[69]
Li X H, Chen J S, Wang X, et al. J. Am. Chem. Soc., 2011, 133:8074-8077 doi: 10.1021/ja200997a
-
[70]
Li X H, Antonietti M. Chem. Soc. Rev., 2013, 42:6593-6604 doi: 10.1039/c3cs60067j
-
[71]
Li X H, Wang X, Antonietti M. Chem. Sci., 2012, 3:2170-2174 doi: 10.1039/c2sc20289a
-
[72]
Lu X, Xu K, Tao S, et al. Chem. Sci., 2016, 7:1462-1467 doi: 10.1039/C5SC03551A
-
[73]
Jorge A B, Martin D J, Dhanoa M T S, et al. J. Phys. Chem. C, 2013, 117:7178-7185 doi: 10.1021/jp4009338
-
[74]
Liu J, Liu Y, Liu N, et al. Science, 2015, 347:970-974 doi: 10.1126/science.aaa3145
-
[75]
Tian J Q, Liu Q, Asiri A M, et al. Anal. Chem., 2013, 85: 5595-5599 doi: 10.1021/ac400924j
-
[76]
Zhang X L, Zheng C, Guo S S, et al. Anal. Chem., 2014, 86: 3426-3434 doi: 10.1021/ac500336f
-
[77]
Wang Q B, Wang W, Lei J P, et al. Anal. Chem., 2013, 85: 12182-12188 doi: 10.1021/ac403646n
-
[78]
Zhang S, Li J, Zeng M, et al. Nanoscale, 2014, 6:4157-4162 doi: 10.1039/c3nr06744k
-
[79]
Bian J, Li Q, Huang C, et al. Nano Energy, 2015, 15:353-361 doi: 10.1016/j.nanoen.2015.04.012
-
[80]
Li G, Lian Z, Wang W, et al. Nano Energy, 2016, 19:446-454 doi: 10.1016/j.nanoen.2015.10.011
-
[81]
Jayaraman T, Arumugam Raja S, Priya A, et al. New J. Chem., 2015, 39:1367-1374 doi: 10.1039/C4NJ01807A
-
[82]
Zhou X, Jin B, Li L, et al. J. Mater. Chem., 2012, 22:17900-17905 doi: 10.1039/c2jm32686h
-
[83]
Su J, Geng P, Li X, et al. Nanoscale, 2015, 7:16282-16289 doi: 10.1039/C5NR04562B
-
[84]
Hou Y, Zuo F, Dagg A P, et al. Adv. Mater., 2014, 26:5043-5049 doi: 10.1002/adma.201401032
-
[85]
Yin H S, Zhou Y L, Li B C, et al. Sens. Actuators B, 2016, 222:1119-1126 doi: 10.1016/j.snb.2015.08.019
-
[86]
Liu Y, Yan K, Zhang J. ACS Appl. Mater. Interfaces, 2016, 8:28255-28264 doi: 10.1021/acsami.5b08275
-
[87]
Li R Y, Zhang Y, Tu W W, et al. ACS Appl. Mater. Interfaces, 2017, 9:22289-22297 doi: 10.1021/acsami.7b06107
-
[88]
Li X, Zhu L, Zhou Y, et al. Anal. Chem., 2017, 89:2369-2376 doi: 10.1021/acs.analchem.6b04184
-
[89]
Zhuang J, Lai W, Xu M, et al. ACS Appl. Mater. Interfaces, 2015, 7:8330-8338 doi: 10.1021/acsami.5b01923
-
[1]
-

图 1 (a) 三嗪基氮化碳结构; (b)三均三嗪基氮化碳结构; (c) melon基氮化碳结构
Figure 1 (a) Triazine-, (b) Tri-s-triazine-and (c) melon-based carbon nitride

图 2 聚合物氮化碳纳米片和块材的理论计算研究: (a)态密度, (b)块材的价带和导带电子波函数图, (c)纳米片的价带和导带电子波函数图[20]
Figure 2 (a) Calculated density of states (DOS) of the single-layered and bulk polymeric carbon nitride, Kohn-Sham orbitals for the valence and conduction bands of (b) bulk and (c) single-layered polymeric carbon nitride, respectively[20]

图 3 聚合物氮化碳材料的光致发光光谱研究: 300 K下, (a)稳态荧光/磷光光谱, (b)时间分辨荧光/磷光光谱, (c)不同延迟时间的磷光光谱, (d)低温下稳态荧光/磷光光谱及时间分辨磷光光谱[26]
Figure 3 (a) Normalized steady-state prompt fluorescence (PF) and phosphorescence (PH) spectra at 300 K, (b) Time-resolved PH kinetics at 300 K, Inset: time-resolved PF kinetics at 300 K, (c) Normalized steady-state PH spectra at different delay times at 300 K, (d) Normalized steady-state PF and PH spectra at 77 K, Inset: time-resolved PH kinetics at 77 K[26]



图 6 聚合物氮化碳材料的激子过程研究: (a)延迟时间依赖瞬态吸收谱的演化; (b)瞬态吸收动力学衰减; (c)相应的光物理模型; (d)材料中三线态-三线态湮没过程示意图[26]
Figure 6 (a) Representative transient absorption spectra at different probe delays and (b) corresponding kinetic traces at different probing wavelengths for g-C3N4; Schematic illustrations of (c) the photophysical processes and (d) triplet-triplet annihilation in the g-C3N4 system[26]


图 8 结构无序度调控聚合物氮化碳能带结构和载流子行为: (a)高温热处理制备结构扭曲氮化碳样品和(b)理论计算模拟结构扭曲对于氮化碳材料光吸收特性的影响[58]; (c)理论模拟无序结构对于聚合物氮化碳能带结构的影响和(d)具有不同无序度聚合物氮化碳材料的光电流瞬态响应曲线[59]
Figure 8 (a) Schematic illustration of designing distorted polymeric carbon nitride nanosheets and (b) calculated theoretical optical absorption of coplanar and corrugation structures[58]; (c) Kohn-Sham orbitals for the valence and conduction bands of pristine g-C3N4 and structural distorted g-C3N4, respectively; (d) Photocurrent responses of samples with different degrees of distortion[59]

图 9 维度控制来调控聚合物氮化碳材料的载流子行为: (a)液相剥离法制备超薄氮化碳纳米片示意图和(b)聚合物氮化碳纳米片和相应块材的荧光发射谱图[20]; (c)氮化碳量子点的透射图像及尺寸分布及其(d)单光子和(e)双光子荧光成像[60]
Figure 9 (a) Schematic illustration of liquid-exfoliation process from bulk g-C3N4 to corresponding ultrathin nanosheets; (b) Normalized photoluminescence spectra of bulk and ultrathin g-C3N4 nanosheets. Inset: the photographs of ultrathin g-C3N4 nanosheets solution without/with UV light illumination[20]; (c) TEM image and corresponding size distribution of the obtained g-C3N4 quantum dots and (d) One-photon and (e) two-photon luminescence image of g-C3N4 quantum dots dispersed in a glass substrate, respectively[60]

图 10 (a) 氮化碳氧化物的HAADF-STEM图和相应元素成像; 氮化碳氧化物和聚合物氮化碳原材料的(b) C1s X射线光电子能谱; (c)磷光光谱和(d)强度归一化的荧光/磷光光谱[33]
Figure 10 (a) Elemental mapping images of CNO; (b) XPS C1s spectra, (c) phosphorescence (PH) spectra and (d) normalized steady-state prompt fluorescence (PF) and PH spectra of pristine g-C3N4 and CNO, respectively[33]

图 11 溴原子对聚合物氮化碳体系光激发过程的影响: (a)瞬态吸收动力学, (b)时间分辨荧光光谱, (c)光催化产氢性能和(d)单线态氧ESR捕获实验[26]
Figure 11 (a) Representative transient absorption kinetic traces, (b) time-resolved prompt fluorescence kinetics, (c) time-dependent photocatalytic H2 evolution, and (d) TEMP-trapped ESR measurements with/without the addition of bromine atoms[26]

图 12 (a) 有序和无序三均三嗪链的密度泛函理论计算; (b)半结晶三均三嗪链中激子解离和电荷转移示意图; (c)离子辅助溶剂热法合成半结晶氮化碳样品的高分辨透射电镜图像; 半结晶氮化碳及其原材料的(d) 300 K下稳态荧光光谱, (e)稳态延迟荧光光谱, (f) Mott-Schottky曲线[68]
Figure 12 (a) DFT simulations of ordered and disordered heptazine-based chains; (b) Schematic illustration of exciton dissociation and charge transfer in semicrystalline heptazine-based melon; (c) High-resolution electron-microscope image of semicrystalline heptazine-based melon, inset: corresponding Fourier transform pattern; Steady-state and time-resolved (d) fluorescence and delayed fluorescence spectra, (e) Mott-Schottky curves of pristine-HM and SC-HM samples, respectively[68]

图 13 聚合物氮化碳/碳量子点材料的(a)形貌表征; (b)可见光下的光催化产氢和产氧量; (c)波长依赖的水分解量子效率; (d)碳量子点浓度依赖的水分解量子效率[74]
Figure 13 (a) Characterization of the physical structure of CDots-C3N4; (b) Time dependent H2 and O2 production under visible light irradiation of CDots-C3N4; (c) Wavelength dependent quantum efficiency of water splitting of CDots-C3N4; (d) Quantum efficiency for different concentrations of CDots in composite catalyst[74]

图 14 氮化碳氧化物及相应块材的(a)气氛依赖的四甲基联苯胺氧化实验和(b)单线态氧ESR捕获测试[33]; 半结晶氮化碳及相应原材料的(c)超氧自由基ESR捕获测试和(d)苯甲醇氧化实验[68]
Figure 14 (a) Evaluation of TMB oxidation under different gas conditions and (b) ESR spectra for singlet oxygen detection with pristine g-C3N4 and CNO[33]; (c) ESR spectra of pristine-HM and SC-HM samples in the presence of DMPO in methanol and (d) wavelength-dependent benzyl alcohol oxidation with pristine-HM and SC-HM sample, respectively[68]

图 15 (A) 聚合物氮化碳纳米片的细胞膜成像实验[20]; (B)聚合物氮化碳量子点对细胞核特异性成像[60]; (C)聚合物氮化碳纳米片的铜离子检测原理图和(D)不同铜离子浓度下,聚合物氮化碳纳米片的荧光谱图[75]
Figure 15 (A) Confocal fluorescence image and overlay image of bright field and confocal fluorescence image of the HeLa cells incubated with ultrathin g-C3N4 nanosheets[20]; (B) One-photon and two-photon confocal fluorescent images of HepG2 cells co-stained with (a) DiI, FDA, g-C3N4 QDs, and (b) DiI, FDA, DAPI, respectively[60]; (C) Schematic illustration of the g-C3N4 nanosheets-based fluorosensor for Cu2+ and (D) PL spectra of g-C3N4 nanosheets dispersion in the presence of different Cu2+ concentrations[75]

图 16 具有分枝结构三氧化钨/聚合物氮化碳/钴氧化物纳米催化剂的(a)合成示意图及其(b)光电催化水氧化性能[84]; 基于聚合物氮化碳/金纳米颗粒光电极的小分子核糖核酸检测(c)机理图及其(d)光电流响应测试[85]
Figure 16 (a) Synthetic route to the three-dimensionally branched WO3 nanosheet array /C3N4 nanosheets//CoOx, and (b) Variation of photocurrent density versus applied voltage of different samples[84]; (c) MicroRNA-319a detection mechanism with g-C3N4-AuNPs nanohybrids and (d) Time-based photocurrent responses of the biosensor[85]
-


 下载:
下载:















 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 117
- 文章访问数: 9918
- HTML全文浏览量: 2508
 下载:
下载:
