 图1
SEM图(a) WO3, (b) MoS2球, (c) MoS2片, (d) S-MoS2/WO3, (e) F-MoS2/WO3; HRTEM图(f) S-MoS2/WO3, (g) F-MoS2/WO3; (h) MoS2/WO3的EDS图
Figure1.
SEM images of (a) WO3, (b) MoS2 microspheres, (c) MoS2 nanosheets, (d) S-MoS2/WO3, (e) F-MoS2/WO3; HRTEM images of (f) S-MoS2/WO3, (g) F-MoS2/WO3; (h) EDS of WO3/MoS2
图1
SEM图(a) WO3, (b) MoS2球, (c) MoS2片, (d) S-MoS2/WO3, (e) F-MoS2/WO3; HRTEM图(f) S-MoS2/WO3, (g) F-MoS2/WO3; (h) MoS2/WO3的EDS图
Figure1.
SEM images of (a) WO3, (b) MoS2 microspheres, (c) MoS2 nanosheets, (d) S-MoS2/WO3, (e) F-MoS2/WO3; HRTEM images of (f) S-MoS2/WO3, (g) F-MoS2/WO3; (h) EDS of WO3/MoS2
引用本文:
侯静静, 赵清华, 李廷鱼, 胡杰, 李朋伟, 李刚. 球状MoS2/WO3复合半导体制备及其对RhB的光催化性能[J]. 无机化学学报,
2017, 33(9): 1527-1536.
doi:
10.11862/CJIC.2017.211

Citation: HOU Jing-Jing, ZHAO Qing-Hua, LI Ting-Yu, HU Jie, LI Peng-Wei, LI Gang. Spherical MoS2/WO3 Composite Semiconductor:Preparation and Photocatalytic Performance for RhB[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(9): 1527-1536. doi: 10.11862/CJIC.2017.211
Citation: HOU Jing-Jing, ZHAO Qing-Hua, LI Ting-Yu, HU Jie, LI Peng-Wei, LI Gang. Spherical MoS2/WO3 Composite Semiconductor:Preparation and Photocatalytic Performance for RhB[J]. Chinese Journal of Inorganic Chemistry, 2017, 33(9): 1527-1536. doi: 10.11862/CJIC.2017.211
球状MoS2/WO3复合半导体制备及其对RhB的光催化性能
摘要:
采用水热法成功制备了MoS2/WO3复合半导体光催化剂,分别通过SEM、TEM、EDS、XRD、Raman和DRS对催化剂的形貌,组成及结构进行表征,并用BET模型计算比表面积。对比发现球状MoS2/WO3对罗丹明B(RhB)的光降解效率明显高于纯WO3、片状MoS2/WO3复合半导体。针对球状MoS2/WO3复合半导体,分别研究了MoS2不同负载量(0.5%,1%,2%,5%,10%)对RhB光催化降解性能的影响,结果表明MoS2含量为2%时催化效果最佳。同时,研究了溶液的pH值(pH=1,3,6,7,11)对光催化降解反应活性的影响,结果显示pH=6时降解率最高。当催化剂量增加到1 g·L-1时,30 min后RhB降解率达到96.6%。球状MoS2/WO3的瞬态光电流为0.050 6 mA·cm-2,比纯WO3提高了2.4倍。经过5次循环实验,球状MoS2/WO3复合半导体催化剂仍能保持90%的高降解率。
English
Spherical MoS2/WO3 Composite Semiconductor:Preparation and Photocatalytic Performance for RhB
Abstract:
MoS2/WO3 composite photocatalyst was synthesized by a hydrothermal method. The morphology and structure of samples were respectively characterized by SEM, TEM, XRD, Raman, BET and DRS. Compared with pure WO3 and flaky MoS2/WO3, the results show that spherical MoS2/WO3 photocatalysts possess higher photocatalytic efficiency for RhB. For the spherical MoS2/WO3 composite semiconductor, the effect of MoS2 loading content (0.5%, 1%, 2%, 5%, 10%) on the photocatalytic degradation of RhB was investigated. The results show that the catalytic effect is the best when the content of MoS2 is 2%. At the same time, the effect of pH (pH=1, 3, 7, 11) on the photocatalytic activity was studied. The results show that the degradation rate is the highest at pH=6. When the amount of catalyst is 1 g·L-1, the degradation rate of RhB reaches 96.6% after 30 min. The transient photocurrent of spherical MoS2/WO3 is 0.050 6 mA·cm-2, which is increased by a factor of 2.4 compared with that of WO3. After five cycles of stability test, the spherical MoS2/WO3 composite semiconductor catalyst can maintain a high degradation rate of 90%.
-
Key words:
- MoS2/WO3
- / heterojunction
- / photocatalysis
- / degradation
- / RhB
-
0 引言
由于工业的快速发展,有机水污染问题越来越严重。有机污染物较强的毒性、致癌和非生物降解性,对人类及动物的健康造成了很大威胁。半导体光催化是一种高效的环境清洁技术,可以将有机污染物完全分解成对环境无害的物质,如水(H2O)、二氧化碳(CO2)、无机盐等[1-3]。由于反应条件温和、氧化能力强、可直接利用太阳光于常温常压下催化降解废水及空气中的有机污染物,并且具有降解彻底、无二次污染等优点,近年来发展迅速,在环保领域得到了广泛应用。但是,很多半导体光催化剂的带隙不能满足可见光的吸收范围,且电子-空穴对很容易复合,催化效率不高。改善光催化剂性能的方法有多种,如金属离子掺杂[4]、非金属离子掺杂[5]、固溶体[6-7]、表面修饰[8]、复合半导体[9]等。
WO3是一种直接带隙的n型半导体材料,近年来因其物理化学性质稳定、无毒性、耐光腐蚀性良好、电子传输率高等被广泛研究[10-13]。此外,它的带隙较小,约为2.4~2.8 eV,可见光即可激发其催化性能,能够有效地利用太阳光谱。然而,由于纯WO3的导带位置较低,且光生电子-空穴对的复合率高,光催化速率还有待提高。目前,研究者通常通过掺杂来引入杂质能级,有效分离光生电子-空穴对以改善WO3光催化效率。Han等[14]通过非金属S掺杂WO3纳米线,在价带上引入中间能级,显著提高了WO3降解甲基橙的能力。Ramkumar等[15]通过简单的化学浴沉积法合成了金属Ag掺杂的WO3薄膜结构,并证实Ag的加入降低了电荷的复合。但是,掺杂会使半导体的热稳定性降低,产生的缺陷位也会成为载流子的复合中心,而复合半导体不存在这样的问题,而且复合半导体能拓宽催化剂的光谱吸收范围,有效分离光生电子-空穴对,提高光催化性能,目前成为一种有效的解决方法[16-17]。二硫化钼(MoS2)是一种新兴的类石墨烯层状化合物,带宽的变化范围为1.2~1.95 eV,每个单元是S-Mo-S的“三明治”结构,其Mo-S棱面相当多,层内以共价键紧密结合在一起,而层间以微弱的范德华力连接[18-20]。这种独特结构使MoS2拥有很多优异的性能,如带隙可调、化学性质稳定、比表面积大、层状边缘有大量的不饱和活性位点等[21-22]。在催化性能方面,一方面大量的不饱和活性位点使MoS2有较高的吸附能力,有助于提高反应活性,如Zhang等[23]通过水热法制备出了MoS2/Bi2WO6核-壳异质结构来降解亚甲基蓝,MoS2的加入明显增强了催化剂的吸附能力,且MoS2/Bi2WO6的光催化活性相比Bi2WO6提高了82%;另一方面,稳定的化学性质、大量的Mo-S棱面使MoS2有大的比表面积都是光催化领域研究者专注于解决的问题,如Awasthi等[24]通过水热法将ZnO纳米花负载到高比表面积的MoS2片上来降解污染物苯酚红,其催化效率和稳定性显著高于同等条件下ZnO和MoS2。因此,半导体中掺入MoS2可以有效地促进光催化活性。另外,MoS2的导带位置比WO3高,二者结合形成异质结可有效解决WO3导带位置低的问题。此外,半导体的形貌结构对光催化效率也有重要影响,纳米MoS2有多种形貌,比如纳米片[25],纳米管[26],纳米线[27],纳米棒[28],纳米球[29],纳米花[30]等,其中,MoS2纳米片和纳米球制备方法简单,比表面积较高,研究最为常见。
本文首先分别采用超声剥离和水热反应法制备出了两种不同形貌的MoS2(纳米片、纳米球),然后采用水热反应将WO3分别和MoS2纳米片、MoS2纳米球相结合形成异质结,成功制备出了两种形貌的MoS2/WO3复合半导体(简记为F-MoS2/WO3和S-MoS2/WO3),并将其用于光催化降解污染物,提高了太阳能利用率和电荷传输效率。以RhB为待测有机污染物,测试并得出了不同的MoS2形貌、负载量、溶液的pH值、催化剂量以及添加H2O2抑制剂条件下,MoS2/WO3复合半导体在可见光下降解RhB的最佳参数。
1 实验部分
1.1 催化剂制备
1.1.1 球状纳米MoS2制备
以钼酸钠(Na2MoO4·2H2O)和硫脲(CN2H4S)为原材料,水热反应制备MoS2纳米球。首先,将称量好的钼酸钠(0.242 g)和硫脲(0.381 g)溶解在60 mL去离子水中。然后,将混合溶液倒入100 mL的水热反应釜中,210 ℃加热24 h得到黑色样品。分别用去离子水和无水乙醇对样品洗涤3次,随后转移至干燥箱,在50 ℃的温度下干燥6 h,最终得到黑色MoS2纳米球粉末。
1.1.2 片状纳米MoS2制备
以块状MoS2粉末和N-甲基吡咯烷酮为原料,梯度离心制备MoS2纳米片。将一定量的块状MoS2加入N-甲基吡咯烷酮中(浓度7.5 mg·mL-1),超声处理90 min,然后将得到的悬浮液静置一晚。取上清液以6 000 r·min-1的转速离心处理60 min,再取1/3上清液以13 000 r·min-1的转速离心30 min,取下层沉淀于50 ℃的温度下干燥6 h即得少数层的MoS2纳米片。
1.1.3 复合半导体WO3/MoS2制备
以六氯化钨(WCl6)和制备好的MoS2为原料,水热反应制备MoS2/WO3复合半导体。称取0.5 g WCl6和一定量已制备好的MoS2,依次倒入50 mL无水乙醇中,然后用磁力搅拌器搅拌一段时间。待两种物质混合均匀后,将其倒入100 mL的水热反应釜中,160 ℃反应处理24 h。等反应釜自然冷却至室温,离心得到蓝色絮状样品。清洗样品以去除杂质离子和未反应的原料,将所得样品于80 ℃下干燥8 h。最后,将样品于管式炉中400 ℃下退火处理2 h,研磨备用。改变MoS2的量,分别制备了0.5%、1%、2%、5%、10% MoS2负载的MoS2/WO3复合半导体。
1.2 催化剂的表征
SEM采用日本的日立SU-3500,加速电压为15 kV,HRTEM采用JEM-2010高分辨透射电子显微镜,加速电压为200 kV。XRD采用辽宁丹东方圆的DX-2007型X射线衍射仪,Cu Kα辐射(λ=0.154 nm),工作电压40 kV,工作电流30 mA,扫描范围10°~90°。Raman测定采用英国的Renishaw InVia-10410拉曼光谱仪,激光波长为514 nm,功率为5 mW。UV-Vis紫外-可见分光光度计采用上海元析UV-8000A来检测样品的吸收谱,扫描范围为190~ 1 100 nm。DRS紫外-可见漫反射吸收光谱采用日本SHIMADZU的UV-2700,BaSO4作为参比。N2吸附-脱吸附采用北京金埃谱的V-Sorb 2800P型物理吸附脱附仪进行测试,200 ℃下真空脱气。光电流采用德国ZAHNER的Zennium电化学工作站测试。
1.3 光催化降解实验
光催化降解实验采用300 W卤钨灯作为光源模拟可见光,光源与待测溶液相距50 cm,并采用自制的冷却系统保持溶液恒温,RhB作为待测污染物。配制10 mg·L-1的RhB溶液,称取0.01 g合成的光催化剂加入100 mL上述溶液中。为了保证光催化剂和污染物之间的吸附-解吸附平衡,上述混合物先在黑暗处避光搅拌30 min。然后,将溶液置于光照下,每隔30 min取一次样,离心处理后用紫外可见分光光度计(UV-vis spectrometer)测溶液的吸收光谱。通过测量RhB溶液在~554 nm处主吸收峰的强度变化来判断溶液的降解量。为确保实验的重复性,所有的光降解实验至少重复3次。光催化效率表示如下:
其中,η为降解率,A0为溶液的初始吸光度,Ai为各个时间段的吸光度。由于溶液的浓度和吸光度成正比,可用吸光度的改变率来计算溶液的降解率。
2 结果与讨论
2.1 微观形貌分析
待测样品的微观形貌结构如图 1所示,由图 1(a~c)可以看出,成功制备出了花状WO3、球状MoS2和少数层片状MoS2。其中,花状WO3是由纳米薄片于不同方向穿插形成的1~2 μm的纳米花,球状MoS2由纳米丝互相缠结而成,比表面积相比片状MoS2要大。如图 1(d)所示,所得S-MoS2/WO3复合半导体中两种物质紧密结合,且MoS2球的加入没有改变球状WO3的形貌。由图 1(e)可以看出,WO3分布在MoS2片表面上,但是MoS2纳米片的添加使得WO3纳米薄片更紧凑。图 1(f~g)分别是S-MoS2/WO3和F-MoS2/WO3的HRTEM图,图(f)可观察到两种晶格条纹,晶面间距0.62和0.38 nm分别对应MoS2和WO3(110) 面;图 1(g)可观察到MoS2和WO3(022) 面的晶格,晶面间距为0.62和0.38 nm。WO3和MoS2之间接触良好,形成的异质结有利于光生电子和空穴在两种半导体材料间迅速迁移,缩短电荷传输距离,实现电子与空穴的高效分离,提高光催化降解RhB的效率。图 1(h)是MoS2/WO3复合半导体的EDS图谱,图中显示催化剂由W、Mo、S、O四种元素组成,无其它杂质元素存在,说明样品较纯。
图1
SEM图(a) WO3, (b) MoS2球, (c) MoS2片, (d) S-MoS2/WO3, (e) F-MoS2/WO3; HRTEM图(f) S-MoS2/WO3, (g) F-MoS2/WO3; (h) MoS2/WO3的EDS图
Figure1.
SEM images of (a) WO3, (b) MoS2 microspheres, (c) MoS2 nanosheets, (d) S-MoS2/WO3, (e) F-MoS2/WO3; HRTEM images of (f) S-MoS2/WO3, (g) F-MoS2/WO3; (h) EDS of WO3/MoS2
2.2 晶相结构和拉曼光谱分析
为了进一步确定催化剂的物相组成,我们测试了WO3和MoS2/WO3复合半导体的XRD图(硅基底衍射峰未标出),结果如图 2(a)所示。与PDF No.43-1035的数据对照,单斜晶体WO3的主峰2θ=23.1°、23.7°、24.4°分别对应于(002)、(020)、(200) 面,并且没有观察到WO3的其他相和WxOy的峰位,这说明我们制备的WO3不含其它杂质。此外,WO3尖锐的峰表明所制备的样品结晶度良好。MoS2/WO3复合半导体的衍射峰中,除了WO3的衍射峰外,还观察到了MoS2的典型特性峰2θ=14.5°((002) 面),这初步表明成功制备出了复合半导体MoS2/WO3。微弱的(002) 面的特征峰证实了复合物中MoS2的比例较低,与实验相符。
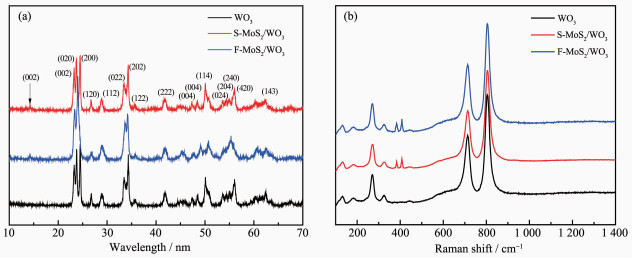 图2
样品的(a) XRD图, (b)拉曼光谱图
Figure2.
(a) XRD patterns and (b) Raman spectra of samples
图2
样品的(a) XRD图, (b)拉曼光谱图
Figure2.
(a) XRD patterns and (b) Raman spectra of samples
如图 2(b)所示,进一步对WO3和MoS2/WO3复合半导体进行拉曼光谱测试。从图 3中可以看出,位于~135、273、713和804 cm-1处的4个特征模式与单斜WO3相吻合。其中,~135、~273 cm-1处的模式与桥接氧的弯曲(O-W-O)振动相关,~713、~806 cm-1处的模式与(W-O)拉伸模式相符合。另外,光谱~383、~408 cm-1处2个散射峰分别与MoS2的拉曼活性振动模式E2g1和A1g相关。E2g1用来表征Mo、S原子的面内振动,而A1g表征的是S原子的面外振动。因此,拉曼光谱测试进一步证明成功合成了MoS2/WO3复合半导体。
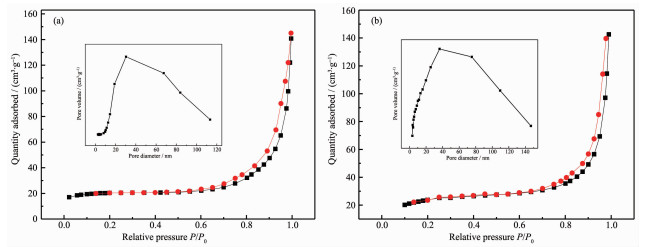 图3
样品的吸附-脱附等温线及孔径分布曲线: (a) S-MoS2/WO3, (b) F-MoS2/WO3
Figure3.
Adsorption-desorption isotherms and pore volume distribution curves of (a) S-MoS2/WO3, (b) F-MoS2/WO3
图3
样品的吸附-脱附等温线及孔径分布曲线: (a) S-MoS2/WO3, (b) F-MoS2/WO3
Figure3.
Adsorption-desorption isotherms and pore volume distribution curves of (a) S-MoS2/WO3, (b) F-MoS2/WO3
2.3 样品的比表面积分析
图 3是S-MoS2/WO3和F-MoS2/WO3的氮气吸附-脱附等温线和孔径分布曲线。根据图 3(a)吸附-脱附曲线可以看出,两种半导体材料均为Ⅳ型吸附脱附等温线,存在H3型滞后环,属于典型的介孔材料。S-MoS2/WO3滞后环的P/P0范围是0.6~1.0,比表面积为15 m2·g-1;F-MoS2/WO3滞后环的P/P0范围是0.7~1.0,比表面积为6 m2·g-1。图 3(b)为材料的孔径分布图,S-MoS2/WO3和F-MoS2/WO3都有较宽的孔径分布范围,分别在29.8和36.6 nm的区域中呈现出最大孔径分数。因此,S-MoS2/WO3的比表面积比F-MoS2/WO3的大,这可以解释上述MoS2纳米片的添加使WO3纳米薄片的穿插更紧凑,即比表面积减小。
2.4 DRS分析
图 4为样品的紫外-可见漫反射吸收光谱。从图 4(a)可知,所有样品在可见光区域(>470 nm)均有明显吸收。WO3与MoS2复合后,对可见光的吸收明显增强,且S-MoS2/WO3的吸收能力高于F-MoS2/WO3。插图可根据如下方程绘制:
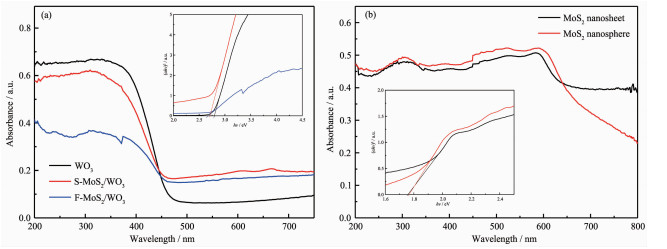 图4
样品的紫外-可见漫反射吸收光谱图: (a) WO3、S-MoS2/WO3和F-MoS2/WO3, (b) MoS2球和MoS2片
Figure4.
UV-Vis diffuse reflectance absorption spectra of (a) WO3, S-MoS2/WO3 and F-MoS2/WO3, (b) MoS2 microspheres and MoS2 nanosheets
图4
样品的紫外-可见漫反射吸收光谱图: (a) WO3、S-MoS2/WO3和F-MoS2/WO3, (b) MoS2球和MoS2片
Figure4.
UV-Vis diffuse reflectance absorption spectra of (a) WO3, S-MoS2/WO3 and F-MoS2/WO3, (b) MoS2 microspheres and MoS2 nanosheets
其中,α,hν,Bi和Eg分别为吸收系数、光子能量、常数和带隙能。WO3、S-MoS2/WO3和F-MoS2/WO3的带隙分别是2.79、2.71、2.61 eV,MoS2的加入降低了WO3的带隙,有助于光催化活性的提高。如图 4(b)所示,球状和片状MoS2对可见光的吸收边界均延伸到了770 nm处,其带隙分别为1.77、1.75 eV。为了确定MoS2/WO3的能带结构,可通过如下公式计算WO3和MoS2的EVB和ECB:
其中,EVB和ECB分别是价带顶和导带底;X是半导体原子电负性的几何平均值,对WO3和MoS2分别为6.59和5.32 eV;Ee是自由电子相对于标准氢电极的能级,约为4.5 eV。经计算,WO3、MoS2球和MoS2片的EVB分别是3.485、1.705和1.695 eV,相应的ECB分别是0.695、-0.065和-0.055 eV,表明WO3和2种形貌的MoS2的带隙均匹配良好,有助于光生电子和空穴的高效分离。
2.5 光催化性能分析
为了对比WO3、F-MoS2/WO3、S-MoS2/WO3的光催化性能,使三者在可见光下分别催化降解RhB溶液。图 5为3种体系降解RhB的UV-Vis吸收谱,该实验在300 W卤钨灯模拟太阳光下进行。催化降解反应中,污染物分子首先被吸附在催化剂的表面,然后再与光生电子和空穴发生一系列氧化还原反应。因此,暗吸附是光催化反应的一个必不可少的环节。暗反应0.5 h后,F-MoS2/WO3的吸附能力高于S-MoS2/WO3,原因可能是片状MoS2结构的裸露表面积更大,即与溶液的接触面积也更大。在前2 h的光照过程中,F-MoS2/WO3的降解率高于S-MoS2/WO3,但随后,S-MoS2/WO3的降解效率明显高于F-MoS2/WO3。可能的原因是:首先,在光照初期,F-MoS2/WO3的裸露表面积大,同时参与降解反应的面积也更大,降解率高;另外,水热合成的MoS2球的层数(约为5~7层)比超声剥离的MoS2片的层数更少,导带电位更高(-0.065 eV),氧化能力增强,且S-MoS2/WO3的比表面积更大(15 m2·g-1),形成的S-MoS2/WO3的活性位点多,光催化活性更高,因此随着反应的进行,最终降解效率高于F-MoS2/WO3。如图 5(a)和(b)所示,降解反应进行到第7 h时,S-MoS2/WO3对RhB降解率达到95%,而F-MoS2/WO3对RhB的降解率只有88%。同时,反应过程中RhB的主吸收峰发生轻微的蓝移(S-MoS2/WO3:554~544 nm,F-MoS2/WO3:554~546 nm)。此外,如图 5(c)所示,在不添加任何催化剂时,RhB的浓度几乎不变,这说明RhB本身不会自动降解,只有在催化剂作用下才能逐步降解。WO3的降解率低于复合半导体MoS2/WO3,表明MoS2的加入可以提高WO3光催化性能。
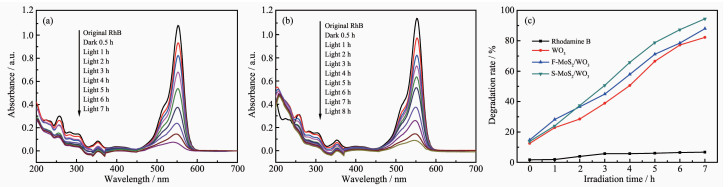 图5
不同阶段的RhB吸收光谱: (a) S-MoS2/WO3, (b) F-MoS2/WO3; (c)降解率随时间的变化
Figure5.
Absorption spectra of RhB solution in different stages: (a) S-MoS2/WO3, (b) F-MoS2/WO3; (c) degradation rate comparison chart
图5
不同阶段的RhB吸收光谱: (a) S-MoS2/WO3, (b) F-MoS2/WO3; (c)降解率随时间的变化
Figure5.
Absorption spectra of RhB solution in different stages: (a) S-MoS2/WO3, (b) F-MoS2/WO3; (c) degradation rate comparison chart
由于S-MoS2/WO3的光催化活性高于F-MoS2/WO3,为了进一步优化S-MoS2/WO3的光催化活性,我们对不同质量比的S-MoS2/WO3复合半导体进行光催化性能测试,如图 6(a)所示。经计算,当反应进行到7 h时,S-MoS2/WO3中MoS2负载量0.5%、1%、2%、5%、10%、20%对应的RhB降解率分别为82%、91%、95%、87%、81%、64%。可以看出,随着MoS2的含量增加,复合半导体的催化效率先增大后减小。当MoS2含量为2%时,催化效率最高,这表明形成的异质结促进了电子-空穴对的分离,提高了光催化活性。当MoS2的含量过多时,额外的MoS2会堆叠在WO3表面,阻碍WO3与溶液的接触,反而降低了催化效率。这说明质量比对复合半导体的催化效率有着很大影响。(以下实验MoS2负载量均为2%)
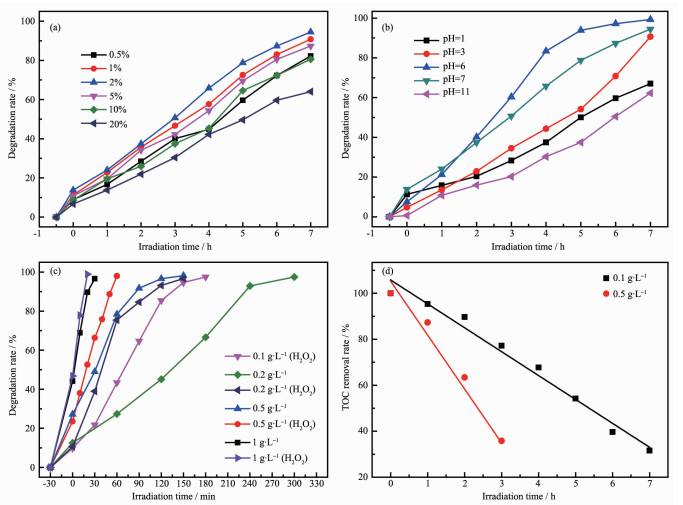 图6
S-MoS2/WO3对RhB的光降解对比图: (a)不同MoS2负载量, (b)不同pH值, (c)不同催化剂量及有无H2O2; (d)溶液的TOC与光照时间变化曲线
Figure6.
Comparison of photocatalytic degradation of RhB by S-MoS2/WO3: (a) different MoS2 loading content, (b) different pH value, (c) different amount of catalyst and whether or not H2O2; (d) temporal changes in total organic carbon (TOC)
图6
S-MoS2/WO3对RhB的光降解对比图: (a)不同MoS2负载量, (b)不同pH值, (c)不同催化剂量及有无H2O2; (d)溶液的TOC与光照时间变化曲线
Figure6.
Comparison of photocatalytic degradation of RhB by S-MoS2/WO3: (a) different MoS2 loading content, (b) different pH value, (c) different amount of catalyst and whether or not H2O2; (d) temporal changes in total organic carbon (TOC)
污染物溶液的pH值对光催化降解效率有一定的影响[31],pH值直接影响催化剂表面所带电荷的性质和污染物在催化剂表面上的吸附行为。通过添加HCl和NaOH配制了不同pH值的RhB溶液,S-MoS2/WO3的光降解对比如图 6(b)所示。随着pH值的提高,S-MoS2/WO3对RhB溶液的降解率呈先上升后下降的趋势,在pH=6时降解率最快。RhB分子本质上带正电,当溶液的pH值大于催化剂的零点电荷pH值时,催化剂表面带负电,可以吸附RhB分子,这有助于溶液的降解。反之,催化剂表面带正电,由于静电斥力作用,RhB分子很难被吸附到催化剂表面,阻碍了反应的进行[32]。由以上分析可得,催化剂S-MoS2/WO3的零点电荷pH值可能小于6。据报道,碱性条件下WO3容易分解[29],方程如下所示:
所以,催化剂在碱性环境下不稳定,pH>7时RhB溶液降解率呈下降趋势。
与已报道的光催化降解RhB的其它催化剂[33-36]及WO3降解其他污染物[37-39]相比,在相同量的污染物浓度下,由于上述所用催化剂量0.1 g·L-1较少,致使催化时间较长,因此图 6(c)测试了不同催化剂量下的催化性能。为了进一步抑制电子-空穴对的复合,暗反应之后在RhB溶液中添加H2O2(100 μL,30%)抑制剂,并比较不添加与添加H2O2催化性能的差异。如图 6(c)所示,催化剂浓度为0.1、0.2、0.5、1 g·L-1的降解率分别是95.3%(420 min)、97.5%(300 min)、98.1%(150 min)、96.6%(30 min)。当催化剂量为1 g·L-1时,降解速率非常快,且暗吸附的作用很大。当催化剂量继续增加至1.5 g·L-1时,暗反应阶段离心后的污染物降解率达到86.4%,由此可见当催化剂量很大时,吸附作用占主导地位。与不含H2O2对比,加入H2O2后降解率分别是97.5%(0.1 g·L-1, 180 min)、96.6%(0.2 g·L-1,150 min)、98.0%(0.5 g·L-1, 60 min)、97.9%(1 g·L-1,20 min),而在暗反应阶段H2O2并没有增加降解率。所以在光照条件下,H2O2可以在很大程度上抑制电子-空穴对的复合,提高光催化效率。
为了验证光催化反应过程中有机碳的浓度是否减小,进行了TOC测试。有机碳的完全消失将证实RhB的完全降解,但若只有少量的减小则可能是光漂白。图 6(d)为溶液的TOC与光照时间曲线,催化剂浓度选取0.1、0.5 g·L-1。随着光照时间的延长,TOC含量不断减少,趋势与RhB降解曲线一致。光催化7 h、3 h后,UV-Vis谱测得RhB完全消失,TOC的浓度分别减少了约70%和65%,表明RhB在S-MoS2/WO3作用下逐渐降解。此外,图 5(a)和(b)中UV-Vis测试的RhB主吸收峰逐渐下降并发生了轻微蓝移,与已报道的研究结果相符[40-41],说明光催化降解RhB过程中“共轭生色团结构分裂”和“N-脱乙基”两种氧化都存在。其中,主吸收峰的快速下降可说明共轭生色团结构分裂占主导地位,主吸收峰的轻微蓝移说明N-脱乙基是次要的。整个降解过程中两种氧化同时进行,先降解为大分子有机物,最后降解为无色小分子化合物, 一部分继续矿化成CO2和H2O[40-43]。
图 7是不同光催化剂的表观一阶动力学曲线,满足以下方程:
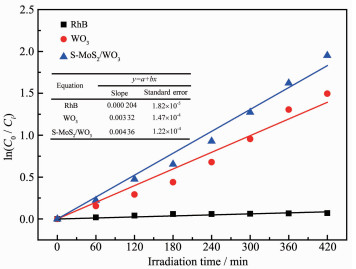 图7
WO3和S-MoS2/WO3降解RhB的动力学曲线
Figure7.
Kinetic curves of RhB degradation for WO3 and S-MoS2/WO3
图7
WO3和S-MoS2/WO3降解RhB的动力学曲线
Figure7.
Kinetic curves of RhB degradation for WO3 and S-MoS2/WO3
其中,C0为RhB溶液暗反应之后的浓度,Ci为反应不同阶段的溶液浓度,kapp是表观速率常数,t为反应时间。kapp和t之间的线性关系说明光催化降解反应遵循表观一阶动力学规律。由图 7中的图表可得,RhB(空白对比试验)、WO3、S-MoS2/WO3的表观速率常数kapp分别是0.000 204、0.003 32、0.004 36 min-1,大小依次为:RhB<WO3<S-MoS2/WO3,即S-MoS2/WO3复合半导体的光催化性能高于纯WO3,这主要是因为复合半导体之间形成的异质结促使光生电子和空穴有效分离,在很大程度上降低了载流子的复合率,提高了其光催化活性[44]。
图 8是WO3和S-MoS2/WO3的光电流对比图,光电化学性能测试是在三电极光电化学池中进行,三电极与电化学工作站相连,光源采用300 W的卤钨灯,电解液为含25%无水甲醇的Na2SO4溶液(0.1 mol·L-1)。工作电极通过滴涂法制备,将4 mg催化剂粉末加入到0.5 mL去离子水中,超声处理10 min,得悬浮液备用。用移液枪移取200 μL悬浮液滴到已处理干净的ITO导电玻璃上,烘干后安装到光电化学池中作为工作电极。由图可知,无光照条件下,催化剂电极的暗态电流趋近于零。给光照瞬间,电流迅速增大,无光照后电流又急剧下降至零,表明本实验所制备的WO3和S-MoS2/WO3二者光电响应速度都很快,具有显著的光电转换特性。在相同条件下,WO3和S-MoS2/WO3的光电流密度分别是0.021 3、0.050 6 mA·cm-2,MoS2球的加入使光电流有很大的提高,表明MoS2球增强了催化剂对可见光的吸收能力,提高了光能利用效率,产生了更多有效传递的电子。因此,S-MoS2/WO3复合半导体表现出了更优越的光催化性能。
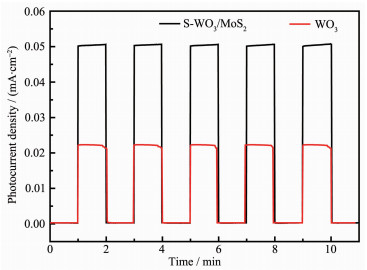 图8
WO3和S-MoS2/WO3的瞬态光电流响应
Figure8.
Transient photocurrent response of WO3 and S-MoS2/WO3
图8
WO3和S-MoS2/WO3的瞬态光电流响应
Figure8.
Transient photocurrent response of WO3 and S-MoS2/WO3
2.6 球状MoS2/WO3异质结的光催化原理
WO3和MoS2的能带结构交错,二者的复合物属于Ⅱ型异质结[31],这种结构为高效的电荷分离提供了最佳的能带位置,催化剂的光催化原理如图 9所示。电子在模拟太阳光的激发下发生跃迁,分别在半导体表面形成电子-空穴对。对于S-MoS2/WO3异质结来说,在由能带相对位置形成的电势差或者内建电场的驱使下,MoS2导带上的光生电子很容易转移到WO3导带上,与此同时WO3价带上的空穴转移到MoS2价带上,使电子和空穴在空间上有效分离。光生电子-空穴对到达半导体表面后发生两类反应:第一类是电子与空穴的简单复合,使吸收的光能以热能的形式散发掉,这类反应由于异质结的加入得到了很大程度上的抑制,即电子-空穴对的复合率很低;第二类是一系列光催化氧化还原反应,污染物的降解即是以此为主要反应,反应细节简单表示如下:
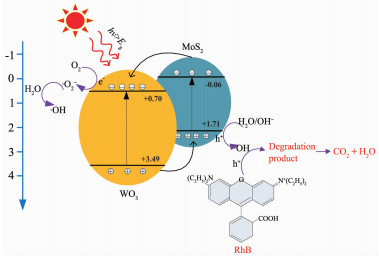 图9
MoS2/WO3光催化活性增强机理
Figure9.
Proposed mechanism for the enhanced photocatalytic activity of MoS2/WO3
图9
MoS2/WO3光催化活性增强机理
Figure9.
Proposed mechanism for the enhanced photocatalytic activity of MoS2/WO3
电子到达WO3表面后吸附氧气,产生超氧基团·O2-,这种超氧基团·O2-和MoS2表面的空穴都能与水反应产生羟基·OH,且H2O2在光照下也能产生·OH。羟基·OH具有很强的氧化性,是污染物降解的核心物质,可将溶液中的有机大分子污染物降解为有机小分子,最后再降解为对环境无害的物质CO2和H2O。另外,空穴也具有强氧化性,与·OH的作用类似,能够直接将污染物氧化。这进一步说明异质结的加入可以促进电子-空穴对的分离,显著降低载流子的复合几率,提高光催化活性。
2.6 光催化剂的稳定性
光催化剂的稳定性是其投入实际应用的一个重要判断因素。因此,S-MoS2/WO3催化性能的稳定性测试非常有必要。经过5次循环测试,降解率如图 10所示。试验中,在每次循环后,离心收集催化剂,用去离子水和无水乙醇各清洗2次,干燥。每次循环实验进行2次或3次,以确保实验后离心收集的催化剂量充足,以备下一次循环测试使用。可以看出,经过5次循环测试后,降解率仅下降了5%,S-MoS2/WO3的光催化稳定性较好。
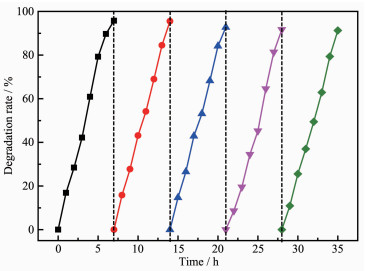 图10
S-MoS2/WO3的循环降解曲线
Figure10.
Recycling degradation curve for S-MoS2/WO3
图10
S-MoS2/WO3的循环降解曲线
Figure10.
Recycling degradation curve for S-MoS2/WO3
3 结论
本文采用水热反应制备出了具有良好光催化效果的MoS2/WO3复合光催化剂,并分别通过SEM、HRTEM、EDS、XRD、Raman、BET和DRS对比两种复合半导体的形貌结构等参数。以RhB为目标污染物,研究了不同实验条件下的光催化效率,结果如下:
(1) 不同催化剂的对比实验显示S-MoS2/WO3对RhB的降解率要明显高于WO3和F-MoS2/WO3,表明MoS2的加入提供了电荷分离场所,提高了催化活性,且S-MoS2/WO3的催化性能更好。
(2) MoS2的不同负载量对S-MoS2/WO3的降解效率有很大影响,研究发现加入的MoS2应适量,过多过少都会影响S-MoS2/WO3的性能,并且结果显示当MoS2所占比例为2%时效率最高,7 h后降解率达到95%。
(3) 溶液的pH值对S-MoS2/WO3的降解效率也有影响,这主要是由于静电影响催化剂与污染物分子之间的吸附作用,结果表明当pH=6时催化剂的效率最高。
(4) 当催化剂量增加时,催化效率有大幅度提高,1 g·L-1的催化剂30 min后降解率达到96.6%。
(5) S-MoS2/WO3的5次循环实验表明该催化剂具有较好的稳定性,可用于污染物的大规模处理。
-
-
[1]
Marschall R. Adv. Funct. Mater., 2014, 24(17):2421-2440 doi: 10.1002/adfm.201303214
-
[2]
Zhang Y H, Chen Z, Liu S Q, et al. Appl. Catal., B, 2013, 140-141:598-607 doi: 10.1016/j.apcatb.2013.04.059
-
[3]
蓝奔月, 史海峰.物理化学学报, 2014, 30(12):2177-2196 doi: 10.3866/PKU.WHXB201409303LAN Ben-Yue, SHI Hai-Feng. Acta Phys. -Chim. Sin., 2014, 30(12):2177-2196 doi: 10.3866/PKU.WHXB201409303
-
[4]
Subash B, Krishnakumar B, Swaminathan M, et al. Mater. Chem. Phys., 2013, 141(1):114-120 doi: 10.1016/j.matchemphys.2013.04.033
-
[5]
Zhao K, Wu Z M, Tang R, et al. J. Korean Chem. Soc., 2013, 57(4):489-492 doi: 10.5012/jkcs.2013.57.4.489
-
[6]
Yang M, Huang Q, Jin X Q. Solid State Sci., 2012, 14(4):465-470 doi: 10.1016/j.solidstatesciences.2012.01.029
-
[7]
Ye L, Han C Q, Ma Z Y, et al. Chem. Eng. J., 2017, 307(1):311-318 http://www.sciencedirect.com/science/article/pii/S1385894716311846
-
[8]
Chakraborty A K, Rawal S B, Han S Y, et al. Appl. Catal., A, 2011, 407(1/2):217-223 http://www.sciencedirect.com/science/article/pii/S0926860X11005072
-
[9]
杜欢, 王晟, 刘恋恋, 等.物理化学学报, 2010, 26(10):2726-2732 doi: 10.3866/PKU.WHXB20101023DU Huan, WANG Sheng, LIU Lian-Lian, et al. Acta Phys.-Chim. Sin., 2010, 26(10):2726-2732 doi: 10.3866/PKU.WHXB20101023
-
[10]
Yao S Y, Zhang X, Qu F Y, et al. J. Alloys Compd., 2016, 689:570-574 doi: 10.1016/j.jallcom.2016.08.025
-
[11]
Mohite S V, Ganbavle V V, Rajpure K Y. J. Alloys Compd., 2016, 655:106-113 doi: 10.1016/j.jallcom.2015.09.154
-
[12]
Srinivasan A, Miyauchi M. J. Phys. Chem. C, 2012, 116(29):15421-15426 doi: 10.1021/jp303472p
-
[13]
刘伯雄, 王金淑, 李鸿义, 等.无机化学学报, 2012, 28(3):465-470 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20120306&journal_id=wjhxxbcnLIU Bai-Xiong, WANG Jing-Shu, LI Hong-Yi, et al. Chinese J. Inorg. Chem., 2012, 28(3):465-470 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20120306&journal_id=wjhxxbcn
-
[14]
Han F G, Li H P, Li F, et al. Chem. Phys. Lett., 2016, 651:183-187 doi: 10.1016/j.cplett.2016.03.017
-
[15]
Ramkumarl S, Rajarajan G. J. Mater. Sci.-Mater. Electron., 2016, 27(11):12185-12192 doi: 10.1007/s10854-016-5373-9
-
[16]
Lu J S, Wang Y J, Liu F, et al. Appl. Surf. Sci., 2017, 393:180-190 doi: 10.1016/j.apsusc.2016.10.003
-
[17]
Du Y, Tang D D, Zhang G K, et al. Chin. J. Catal., 2015, 36(12):2219-2228 doi: 10.1016/S1872-2067(15)61015-4
-
[18]
Li J, Liu E Z, Ma Y N, et al. Appl. Surf. Sci., 2016, 364:694-702 doi: 10.1016/j.apsusc.2015.12.236
-
[19]
Zhou W J, Yin Z Y, Du Y P, et al. Small, 2013, 9(1):140-147 doi: 10.1002/smll.v9.1
-
[20]
Zhao H, Dong Y M, Jiang P P, et al. J. Mater. Chem. A, 2015, 3(14):7375-7381 doi: 10.1039/C5TA00402K
-
[21]
Hu K, Hu X, Xu Y, et al. React. Kinet. Mech. Catal., 2010, 100(1):153-163 doi: 10.1007/s11144-010-0173-3
-
[22]
翟英娇, 李金华, 楚学影, 等.无机材料学报, 2015, 30(9):897-905 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmZHAI Ying-Jiao, LI Jin-Hua, CHU Xue-Ying, et al. J. Inorg. Mater., 2015, 30(9):897-905 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[23]
Zhang J, Huang L H, Jin H Y, et al. Mater. Res. Bull., 2017, 85:140-146 doi: 10.1016/j.materresbull.2016.09.013
-
[24]
Awasthi G P, Adhikari S P, Ko S. J. Alloys Compd., 2016, 682:208-215 doi: 10.1016/j.jallcom.2016.04.267
-
[25]
何亚飞, 郝立峰, 陆小龙, 等.高分子学报, 2015(2):197-203 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmHE Ya-Fei, HAO Li-Feng, LU Xiao-Long, et al. Acta Polym. Sin., 2015(2):197-203 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[26]
Afanasiev P, Bezverkhy I. J. Phys. Chem. B, 2003, 107(12):2678-2683 doi: 10.1021/jp021655k
-
[27]
Li W J, Shi E W, Ko J M, et al. J. Cryst. Growth, 2003, 250(3/4):418-422 http://www.sciencedirect.com/science/article/pii/S0022024802024120
-
[28]
Thamankar R, Yap T L, Goh K E J, et al. Appl. Phys. Lett., 2013, 103(8):83106 doi: 10.1063/1.4818998
-
[29]
Xiang Q J, Yu J G, Jaroniec M. J. Am. Chem. Soc., 2012, 134(15):6575-6578 doi: 10.1021/ja302846n
-
[30]
Anderson C, Bard A J. J. Phys. Chem., 1995, 99(24):9882-9885 doi: 10.1021/j100024a033
-
[31]
Guo Y P, Zhao J Z, Zhang H, et al. Dyes Pigm., 2005, 66(2):123-128 doi: 10.1016/j.dyepig.2004.09.014
-
[32]
方世杰, 徐明霞, 黄卫友, 等.硅酸盐学报, 2001, 29(5):439-442 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmFANG Shi-Jie, XU Ming-Xia, HUANG Wei-You, et al. J. Chin. Ceram. Soc., 2001, 29(5):439-442 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[33]
侯不唯, 李恺.无机化学学报, 2017, 33(6):1007-1014 doi: 10.11862/CJIC.2017.110HOU Bu-Wei, LI Kai. Chinese J. Inorg. Chem., 2017, 33(6):1007-1014 doi: 10.11862/CJIC.2017.110
-
[34]
徐梦秋, 柴波, 闫俊涛, 等.无机化学学报, 2017, 33(3):389-395 doi: 10.11862/CJIC.2017.045XU Meng-Qiu, CHAI Bo, YAN Jun-Tao, et al. Chinese J. Inorg. Chem., 2017, 33(3):389-395 doi: 10.11862/CJIC.2017.045
-
[35]
高志慧, 张秀芳, 马春.大连工业大学学报, 2016, 35(6):441-444 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htmGAO Zhi-Hui, ZHANG Xiu-Fang, MA Chun. J. Dalian Polytech. Univ., 2016, 35(6):441-444 http://www.cnki.com.cn/Article/CJFDTOTAL-SYQY201603027.htm
-
[36]
张田, 邹正光, 何金云, 等.无机化学学报, 2017, 33(6):954-962 doi: 10.11862/CJIC.2017.093ZHANG Tian, ZOU Zheng-Guang, HE Jin-Yun, et al. Chinese J. Inorg. Chem., 2017, 33(6):954-962 doi: 10.11862/CJIC.2017.093
-
[37]
刘柏雄, 王金淑, 李洪义, 等.无机化学学报, 2012, 28(3):465-470 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20120306&journal_id=wjhxxbcnLIU Bo-Xiong, WANG Jin-Shu, LI Hong-Yi, et al. Chinese J. Inorg. Chem., 2012, 28(3):465-470 http://www.wjhxxb.cn/wjhxxbcn/ch/reader/view_abstract.aspx?flag=1&file_no=20120306&journal_id=wjhxxbcn
-
[38]
Hai G J, Huang J F, Cao L Y, et al. J. Alloys Compd., 2017, 690:239-248 doi: 10.1016/j.jallcom.2016.08.099
-
[39]
Chen H H, Xiong X Q, Hao L L, et al. Appl. Surf. Sci., 2016, 389:491-495 doi: 10.1016/j.apsusc.2016.07.145
-
[40]
Yu K, Yang S G, He H, et al. J. Phys. Chem. A, 2009, 113(37):10024-10032 doi: 10.1021/jp905173e
-
[41]
Li X X, Wan T, Qiu J Y, et al. Appl. Catal., B, 2017, 217:591-602 doi: 10.1016/j.apcatb.2017.05.086
-
[42]
Zhang F W, Wen Q J, Hong M Z, et al. Chem. Eng. J., 2017, 307:593-603 doi: 10.1016/j.cej.2016.08.120
-
[43]
Liu W W, Shang Y Y, Zhu A Q, et al. J. Mater. Chem. A, 2013, 5(24):12542-12549 doi: 10.1186/2045-9769-2-7
-
[44]
Matos J, Laine J, Herrmann J M. Appl. Catal., B, 1998, 18(3/4):281-291 http://www.sciencedirect.com/science/article/pii/S0926337398000514
-
[1]
-

图 1 SEM图(a) WO3, (b) MoS2球, (c) MoS2片, (d) S-MoS2/WO3, (e) F-MoS2/WO3; HRTEM图(f) S-MoS2/WO3, (g) F-MoS2/WO3; (h) MoS2/WO3的EDS图
Figure 1 SEM images of (a) WO3, (b) MoS2 microspheres, (c) MoS2 nanosheets, (d) S-MoS2/WO3, (e) F-MoS2/WO3; HRTEM images of (f) S-MoS2/WO3, (g) F-MoS2/WO3; (h) EDS of WO3/MoS2

图 3 样品的吸附-脱附等温线及孔径分布曲线: (a) S-MoS2/WO3, (b) F-MoS2/WO3
Figure 3 Adsorption-desorption isotherms and pore volume distribution curves of (a) S-MoS2/WO3, (b) F-MoS2/WO3

图 4 样品的紫外-可见漫反射吸收光谱图: (a) WO3、S-MoS2/WO3和F-MoS2/WO3, (b) MoS2球和MoS2片
Figure 4 UV-Vis diffuse reflectance absorption spectra of (a) WO3, S-MoS2/WO3 and F-MoS2/WO3, (b) MoS2 microspheres and MoS2 nanosheets

图 5 不同阶段的RhB吸收光谱: (a) S-MoS2/WO3, (b) F-MoS2/WO3; (c)降解率随时间的变化
Figure 5 Absorption spectra of RhB solution in different stages: (a) S-MoS2/WO3, (b) F-MoS2/WO3; (c) degradation rate comparison chart

图 6 S-MoS2/WO3对RhB的光降解对比图: (a)不同MoS2负载量, (b)不同pH值, (c)不同催化剂量及有无H2O2; (d)溶液的TOC与光照时间变化曲线
Figure 6 Comparison of photocatalytic degradation of RhB by S-MoS2/WO3: (a) different MoS2 loading content, (b) different pH value, (c) different amount of catalyst and whether or not H2O2; (d) temporal changes in total organic carbon (TOC)

图 7 WO3和S-MoS2/WO3降解RhB的动力学曲线
Figure 7 Kinetic curves of RhB degradation for WO3 and S-MoS2/WO3

图 8 WO3和S-MoS2/WO3的瞬态光电流响应
Figure 8 Transient photocurrent response of WO3 and S-MoS2/WO3

图 9 MoS2/WO3光催化活性增强机理
Figure 9 Proposed mechanism for the enhanced photocatalytic activity of MoS2/WO3
-


 下载:
下载:









 扫一扫看文章
扫一扫看文章
计量
- PDF下载量: 22
- 文章访问数: 2796
- HTML全文浏览量: 1012
 下载:
下载:
